Pathogenesis and Clinical Features
APS is an autoimmune disease in which antibodies directed against phospholipid-protein complexes appear to lead directly to thrombosis. Like all autoimmune diseases APS has a genetic component, and cases of thyroid disease, Sjögren’s syndrome, and lupus are frequently seen in families with APS.
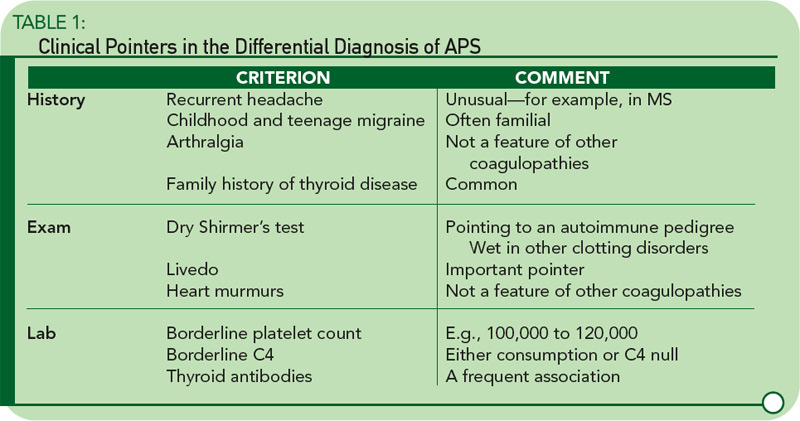
While arterial and venous thromboses are the hallmarks of the syndrome, there are other clinical clues. Headache—particularly migraine—is often an early feature going back to childhood, and often familial. Livedo reticularis is an important physical sign, and a dry Shirmer’s test, suggesting underlying Sjögren’s syndrome, is a clue to the disease’s autoimmune pedigree. Table 1 lists some of the major differences between APS and other pro-thrombotic disorders.
Neurological Considerations
Stroke: The most feared complication of APS is stroke, with up to 20% of strokes in people under 45 possibly having this etiology. In this setting, the MRI can be misleading (e.g., false negatives) and we look forward to the development of more sensitive and available screening techniques. Estimates for the frequency of stroke in patients with APS vary widely, and some studies have been hampered by methodological problems.4