Sarah2 / shutterstock.com
CHICAGO—Ken Smith, MD, PhD, professor of medicine at the University of Cambridge, England, gave an update on anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) at the 2018 ACR/ARHP Annual Meeting.
Although vasculitis tends to be defined first by vessel size, the clinical differentiation between the forms is not reliable, explained Dr. Smith. For example, granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are often treated as a single disease in clinical trials, but in truth they have different clinical features and two different autoantibody reactivities that correlate well, but not perfectly, with classification as GPA or MPA.
Clinicians have long noticed that patients with AAV exhibited familial clustering, like that seen with rheumatoid arthritis (RA). These observations led to the identification of many candidate genes, and genome-wide association studies (GWAS) have allowed for the statistical association between disease and single nucleotide polymorphisms (SNPs). These results, published in 2012, confirmed that AAV has a genetic component.1
The researchers also found genetic distinctions between GPA and MPA that are more strongly associated with ANCA specificity than with the clinical syndrome and imply the response to the autoantigen proteinase 3 (PR3) is a central pathogenic feature of PR3-ANCA-associated vasculitis. This study that provided preliminary support for the hypothesis that PR3-ANCA-associated vasculitis and myeloperoxidase (MPO)-ANCA-associated vasculitis are distinct autoimmune syndromes.
New Data
The recognition of the significant genetic contribution to AAV prompted a more detailed analysis, which yielded three main variants: HLA class II, SERPINA1 and PRTN3, each of which was specifically associated with PR3-ANCA.2 Both the HLA and non-HLA gene variants appear to alter proteins integral to immune responses that likely play key roles in the pathogenesis of AAV.
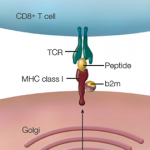
In particular, the largest effect on risk came from a triallellic HLA-DPB1 haplotype, meaning that three gene variants are inherited together. The resulting change in amino acid sequence affects a β-chain polymorphism in the HLA-DP antigen-binding pocket that modifies the protein’s peptide-binding properties and, therefore—possibly—T cell allorecognition. The analyses suggest there is only a single genetic association with the HLA region, and so the other SNPs identified in the region (e.g., collagen) are likely associated by virtue of linkage disequilibrium.
The two other loci related to AAV code for a serine protease inhibitor (serpin), which appears to control key intracellular and extracellular pathways of the auto-antigen PR3.3 New research suggests free PR3 may be more antigenic than complexed PR3, signifying that its role in autoimmunity may be more complicated than originally believed. These nuances can be further explored, because researchers published the genomic atlas of the human plasma proteome, which has expanded the ability to use specific proteins to link genetic factors to diseases.4 The evolving analyses support patient stratification based on ANCA specificity.
Researchers have used the accumulated data to determine why MPO- and PR3-AAV have so many clinical similarities. One hypothesis is that the various forms of ANCA have a common target (neutrophils) and the damage to the neutrophils results in a common clinical manifestation. “It could be a sort of antigenic accident,” explained Dr. Smith. If this is true, then it may be that the balance between costimulatory and coinhibitory signals shape T cell exhaustion and determine whether the immune system becomes autoreactive.5
EGPA
Dr. Smith transitioned to a discussion of unpublished genetic data on the rare disease eosinophilic granulomatosis with polyangiitis (EGPA), also known as the Churg–Strauss syndrome. The features of EGPA that are present in almost all patients are asthma and eosinophilia, and additional classification criteria include paranasal sinus abnormalities, infiltrates on chest imaging, neuropathy and histological evidence of extravascular eosinophils. Patients with EGPA can get very sick if untreated, with eosinophilic myocarditis as a life-threatening feature and neuropathy as a common source of disability.
Because many patients with EGPA first present with asthma symptoms, a GWAS of patients with asthma has revealed some clues to the pathophysiology of EGPA.6 Although asthma is genetically heterogeneous, a few common alleles are associated with disease risk. The implicated genes suggest asthma results from a breakdown in the communication of epithelial damage to the adaptive immune system as well as the activation of airway inflammation. Surprisingly (at the time), elevation of total serum IgE levels had only a minor role in the development of asthma. Instead, variants in the ORMDL3/GSDMB locus were strongly associated only with childhood-onset disease. These results, in combination with a more recent GWAS study that interrogated 36 traits across the hematopoietic system, revealed a genetic relatedness between EGPA and asthma, which was consistent with their shared relatedness to eosinophil count.7
Dr. Smith has now documented 11 EGPA SNP associations that support the hypothesis that EGPA is a polygenic disease. Ten of the 11 EGPA loci are associated with eosinophil count, which suggests a unifying hypothesis in which eosinophil count drives the EGPA prodrome.
Genetic variants associated with long-term outcomes, however, are likely to be distinct from those that drive the susceptibility to disease, and therefore future research efforts will focus on genetics’ role on treatment response, which should make it possible to identify new therapies that target GWAS-identified pathways. Continued analysis of EGPA and other AAV subgroups could yield novel potential therapeutic targets, as well as a path toward individualized therapy.8
Lara C. Pullen, PhD, is a medical writer based in the Chicago area.
References
- Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012 Jul 19;367(3):214–223.
- Merkel PA, Xie G, Monach PA, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody–associated vasculitis. Arthritis Rheumatol. 2017 May;69(5):1054–1066.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med. 2002 Jan 3;346(1):45–53.
- Sun BS, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018 Jun;558(7708):73–79.
- McKinney EF, Lee JC, Jayne DRW, et al. T cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015 Jul 30;523(7652):612–616.
- Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genome wide association study of asthma. N Engl J Med. 2010 Sep 23;363(13):1211–1221.
- Astle WJ, Elding H, Jiang T, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016 Nov 17;167(5):1415–1429.
- McKinney EF, Lyons P, Carr EJ, et al. A CD8 T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010 May;16(5):586–591.