
The acute care of a critically ill patient with systemic lupus erythematosus (SLE) and new neurological symptoms is one of the most challenging dilemmas in rheumatology. Despite an ever-growing array of cutting-edge imaging modalities and other clinical tools, our understanding of central nervous system (CNS) disease in SLE remains incomplete. Fortunately, for a subset of patients with SLE who present with seizures, such as the patient presented herein, we are making some headway, with important implications for treatment.
The Case
The patient is a 28-year-old woman with a two-year history of SLE. She had initially presented with Raynaud’s, chilblains and arthralgias. Labs revealed anemia, leukopenia, an antinuclear antibody (ANA) titer of 1:160, positive anti-double-stranded DNA (dsDNA), and positive myeloperoxidase (MPO) and anti-Ro antibodies. She was treated with hydroxychloroquine 400 mg daily, 50 mg azathioprine daily and 10 mg prednisone daily. She had not tolerated mycophenolate mofetil or mycophenolic acid due to gastrointestinal (GI) side effects.
Her GI illness improved over the next several days. The stress-dose steroids were weaned down to her home dose of prednisone 10 mg daily. Arrangements were made for discharge home on hospital Day 7.
However, early in the morning on her planned day of discharge, she had a generalized tonic clonic seizure. She had never before had a seizure. She was given intravenous lorazepam. A head CT without contrast was unremarkable. During the subsequent eight-hour period, she had an additional three seizures, despite IV leveteracetam and additional doses of IV lorazepam. She was intubated and sedated, and in the early evening, she was transferred to our institution for further evaluation and treatment. The rheumatology team was consulted immediately upon her arrival to help determine whether her seizures represented a manifestation of neuropsychiatric systemic lupus erythematosus (NPSLE).
NPSLE
Despite the best efforts of experts in the field, NPSLE remains vaguely defined. In 1999, an ad hoc committee assembled by the ACR produced formal definitions of 19 NPSLE syndromes, ranging from headache to cerebrovascular disease.1 Unfortunately, although these definitions have been important for providing uniform nomenclature among researchers, numerous other diseases can cause presentations identical to each NPSLE syndrome. Many complications of SLE, such as uremia caused by lupus nephritis, and medications used to treat SLE can themselves cause neurological disease, which could be considered forms of secondary NPSLE. All in all, CNS symptoms in a patient with SLE are often a diagnostic conundrum with a very broad differential diagnosis.
Primary NPSLE is thought to occur via one of two possible types of mechanisms.2 The better understood of these is vascular insult, in which antiphospholipid antibodies or other risk factors for hypercoagulation lead to thrombi and focal regions of ischemia, causing clinical presentations, such as stroke or seizure. There may also be additional small vessel injury in chronic disease resulting in vasculopathy.
The second mechanism, inflammatory NPSLE, is poorly understood. Autopsy studies on patients with SLE have shown that CNS vasculitis is very, very rare. Instead, it is thought that inflammation may be mediated through antineuronal antibodies or cytokine injury (Note: Elevated levels of interleukin 6 in SLE correlate with increased risk of seizures).2
So far, no particular serological marker has proved to be predictive of NPSLE. Presence of antiribosomal P was proposed to be such a marker. Subsequent studies, however, estimate its sensitivity at 26% and specificity at 80%, limiting the utility of this test.3 With the diverse pathogenic mechanisms causing NPSLE, there may not be a single predictive marker. Elucidating the pathophysiology of the many NPSLE syndromes will be crucial in developing a better framework for its diagnosis and treatment.
Diagnosis
Because the possible neuropsychological effects of lupus are broad and can affect both the central and peripheral nervous systems, the diagnostic workup is guided by anatomic localization and clinical suspicion. The workup will uniformly require basic labs—especially because metabolic derangements are a common cause of neurological disease—as well as imaging.
For acute presentations, such as seizures, an immediate head CT without contrast is necessary to rule out hemorrhage or other severe structural disease. For most cases of neuropsychiatric disease in the SLE population, a magnetic resonance imaging (MRI) of the brain will also be indicated. Functional imaging (e.g., positron emission tomography [PET] or functional MRI [fMRI]) has shown promise in recent studies, particularly for cognitive dysfunction, but the findings are not specific for NPSLE.
The initial workup could also include blood cultures and a lumbar puncture for cerebrospinal fluid (CSF) studies to rule out infectious etiologies of neurological disease. Remember, it’s not safe to perform a lumbar puncture in the patient with headache and neurological abnormalities without initial neuroimaging for risk of provoking herniation from abrupt changes in intracranial pressure.
Last, but not least, lab testing should include markers of SLE activity (e.g., dsDNA, complement activity or level, erythrocyte sedimentation rate [ESR], C-reactive protein [CRP]) as well as antiphospholipid antibody testing, even if prior hypercoagulability testing has been negative.
Back to the Case
In rheumatology, a patient’s verbal description of their symptoms and triggers carries great importance, and this is no less true when patients present with neurological symptoms. In this case, with the patient intubated and deeply sedated, we were not able to gather any such information from the patient herself. Fortunately, her mother recalled that the patient had been moderately hypertensive (i.e., systolic BPs as high as the 170s) for several days prior to the seizures and that she had noted a visual disturbance in the form of “a rainbow of colors” immediately before her first seizure.
Labs revealed a serum creatinine at a baseline level of 0.63 mg/dL, stable blood cell and platelet counts, and no antiphospholipid antibodies. Her dsDNA was >1,000 international units, which is her baseline, and her complement levels (C3 and C4) were 27 mg/dL and 4 mg/dL, respectively, compared with her usual levels of 60 mg/dL and 10 mg/dL. ESR and CRP were 16 mm/hour and 6 mg/L, respectively.
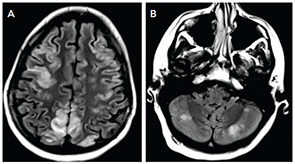
The MRI of the brain showed cortical and subcortical T2 hyperintensities primarily involving bilateral parietal, occipital and posterior temporal lobes (see Figure 1). T2 hyperintense lesions were also seen in the subcortical frontal lobe and bilateral cerebellar hemispheres. There was no evidence of acute ischemic infarct. Lumbar puncture showed three white blood cells, 2,800 red blood cells, glucose of 41 mg/dL and protein of 31 mg/dL. The history of headache, seizure and visual disturbance combined with the MRI findings and noninflammatory CSF are most consistent with posterior reversible encephalopathy syndrome. Levetiracetam for seizure prevention was started, and BP was controlled to systolic of less than 140 mmHg. Over the next three days, her mental status improved to baseline, the headache resolved, and she did not have further seizures or visual disturbances.
PRES
Posterior reversible encephalopathy syndrome (PRES) is characterized by subacute onset of headache, seizure, encephalopathy or visual disturbance, with neuroimaging showing reversible subcortical vasogenic edema (see Table 1). Although PRES was named as such in 1996, the disorder had been known for many years, initially as hypertensive encephalopathy and then as reversible posterior leukoencephalopathy.
Clinically, seizures are the most common mode of presentation, occurring in about 75% or more. Visual disturbance is cortically mediated and can take the form of cortical blindness, visual neglect or higher order visual processing difficulty (e.g., simultagnosia, in which patients can identify parts of a picture but not put together the entire picture—identify the trees, but not the forest). The most common imaging feature on MRI is presence of relatively symmetric T2 hyperintensity, thought to be edema, in the posterior white matter.
Despite having “posterior” in the name, PRES can affect other regions as well, including frontal lobes, basal ganglia, the brainstem and cerebellum, as they did in our patient. Some of these changes may be apparent on head CT, but MRI is more sensitive for this condition.
PRES is associated with hypertension found in the majority of patients (mean systolic BP of about 190 mmHg). In 10% of patients, PRES can occur with systolic BP below 140 mmHg.4 Abrupt increases in BP may be pathogenic even when the absolute systolic pressure is not that high. Hypertension alone, however, does not always cause PRES.

Other common risk factors are acute or chronic renal failure; chemotherapy/immunosuppressive medication, including cyclophosphamide, calcineurin inhibitors and methotrexate; bevacizumab; and eclampsia.
Many patients with PRES have concurrent autoimmune disorders, estimated at 45% in case series, including SLE, rheumatoid arthritis, scleroderma and ulcerative colitis. Some of these conditions, however, are also associated with use of immunosuppressive medications, hypertension or renal dysfunction. The relative contribution of the underlying autoimmune disorder vs. its complications in predisposing to PRES is uncertain. Our patient, for example, had risk factors of preceding renal dysfunction and prior use of immunosuppressive medications.
There is a growing literature of cases of PRES occurring in patients with SLE, some in the absence of renal dysfunction or immunosuppressive medications, suggesting that SLE alone may also be a risk factor.5 Further epidemiologic studies are needed to clarify the relationship.
In PRES, there is dysfunction of cerebral vascular autoregulation and breakdown of the blood–brain barrier. The cause of this dysregulation is unclear, but it’s hypothesized to result from endothelial injury, such as from rapid rise in BP or vasospasm from circulating toxins. So far, there is no evidence of inflammation contributing to PRES.
The treatment is primarily supportive, consisting of treating seizures and hypertension and removing offending medications.
A detailed medication history is helpful to determine the triggering medication because the majority of reactions occur within two weeks of initiation, although in about 20% of cases, PRES from a medication can occur weeks or months later.6 Escalation of chemotherapy/immunosuppressive medications is avoided in the acute phase (e.g., pulse-dose corticosteroids or cyclophosphamide for treating SLE flare). In the majority of patients, symptoms generally resolve in three–eight days.
The MRI can remain abnormal despite improvement in clinical symptoms, although repeat imaging after weeks generally shows resolution of the vasogenic edema. Sometimes, in patients with PRES, there may develop a concurrent intracerebral hemorrhage or ischemic infarction. In these cases, recovery may not be complete.
Patient Outcome
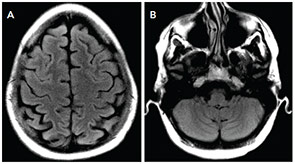
Our patient had complete neurological recovery within days. She has not had any further seizures in the four months since her presentation, and her anti-epileptic medication has been weaned off. (see Figure 2).
In the interim, a second patient was admitted to our hospital with a similar presentation of septic shock followed by seizures in the setting of relative hypertension, with MRI findings consistent with PRES. She, too, recovered fully with supportive care and BP control. Interestingly, she did not have a history of lupus nephritis, although she did have acute renal failure in the setting of septic shock, and she was only on hydroxychloroquine as an outpatient medication.
Conclusion
We suspect that PRES in patients with SLE may be more common than previously suspected, and we hope that both rheumatologists and neurologists will become more aware of this potential diagnosis in patients with SLE who present with seizures or even just headache. Whether or not it should be considered a form of NPSLE remains a matter of debate, but the importance of BP control (not immunosuppression) is clear.
Anna Helena Jonsson, MD, PhD, is a first-year rheumatology fellow at Brigham and Women’s Hospital in Boston.
Shamik Bhattacharyya, MD, is a senior resident in neurology at Massachusetts General Hospital and Brigham and Women’s Hospital in Boston.
References
- ACR Ad Hoc Committee on Neuropsychiatric Lupus Nomenclature. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599–608.
- Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 2014;10(6):338–347.
- Karassa FB, Afeltra A, Ambrozic A, et al. Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2006;54(1):312–324.
- Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: Associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427–432.
- Barber CE, Leclerc R, Gladman DD, et al. Posterior reversible encephalopathy syndrome: An emerging disease manifestation in systemic lupus erythematosus. Semin Arthritis Rheum. 2001;41(3):353–363.
- Roth C, Ferbert A. The posterior reversible encephalopathy syndrome: What’s certain, what’s new? Pract Neurol. 2011;11(3):136–144.
