CNK02 / shutterstock.com
Scleroderma, or systemic sclerosis (SSc), is an autoimmune disease characterized by vasculopathy and fibrosis. Although relatively rare, with a prevalence in North America of approximately 300 per 1 million people, SSc is associated with significant morbidity and high rates of mortality.1 Patients with scleroderma have four times greater mortality than age- and sex-matched controls, with the majority of deaths related to interstitial lung disease (ILD) and pulmonary hypertension (PH).2 Predicting disease course at baseline is, therefore, of great importance, with implications for the choice of therapy. In 2013, the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) developed new classification criteria for SSc.3 These criteria allow for the inclusion of patients with early manifestations of the disease, such as puffy fingers, Raynaud’s phenomenon, nailfold capillary changes and specific auto-antibodies.
Identification of patients in the very early stages of disease is useful in helping establish care with a rheumatologist at a specialized center and for the implementation of treatment at earlier stages.
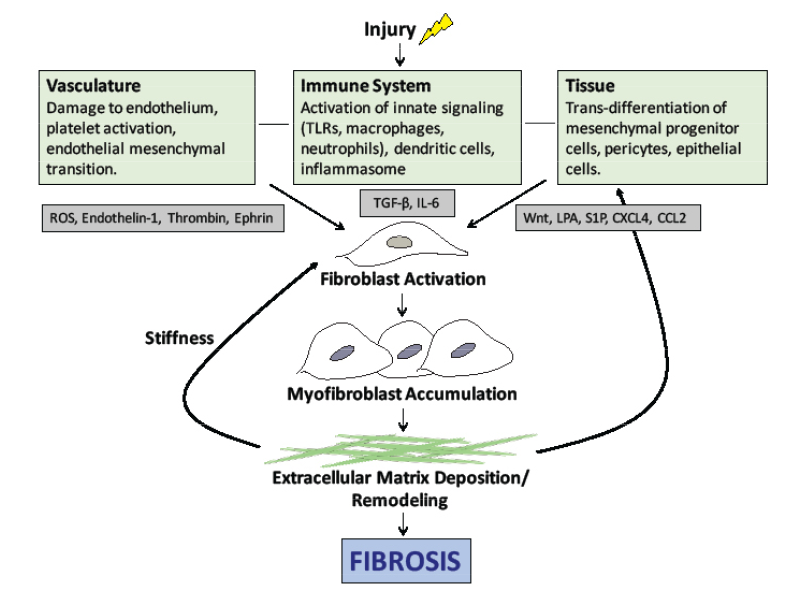
The central mediator of SSc fibrosis is the pleotropic cytokine, transforming growth factor-β (TGF-β). At baseline, TGF-β is in a latent form and must be activated by αv integrins. Active TGF-β then stimulates fibroblasts to proliferate and convert to myofibroblasts, which are responsible for laying down the extracellular matrix (ECM). Interleukin 6 (IL-6) is also secreted by activated immune cells and plays a role in fibroblast accumulation. Previous data suggest IL-6 levels are increased in skin and serum of patients with SSc and that elevated IL-6 levels predict severity of skin involvement in SSc.4
The bioactive lipid mediators, lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P), are also implicated in SSc pathogenesis. Selective deletion of the LPA1 receptor in mouse models of fibrosis leads to a marked decrease in both lung and skin fibrosis.5,6 The interplay of these mediators leads to progressive ECM accumulation and structural remodeling, which causes further activation of fibroblasts and results in the progression of fibrosis.
Effective therapies to prevent or halt this progression will need to disrupt the cellular events illustrated in Figure 1).
Challenges in Scleroderma Treatment
The incomplete understanding of the pathogenesis of scleroderma has historically been one barrier to the development of effective treatments for the disease. However, additional challenges exist in developing new and effective treatments for patients with scleroderma (see Table 1).

(click for larger image)
TABLE 1: Challenges in SSc Clinical Trials
The disease is heterogeneous, with some patients having severe skin involvement only and others having internal organ involvement such as ILD, PH, gastrointestinal disease or renal crisis. This heterogeneity poses challenges when trying to figure out a one-drug-fits-all approach and affects enrollment of patients in clinical trials.
Similarly, there is variability in the trajectory of the disease, with some patients having rapid progression of their disease and others remaining stable for many years. Predicting which patients may have more rapid progression of disease remains a challenge. To date, clinical outcomes in trials have focused on the modified Rodnan skin score (mRSS; a validated measure of skin thickness in 17 locations, graded on a scale of 0–3) and the forced vital capacity (FVC). New efforts are underway to develop a more comprehensive outcome measure that would incorporate multiple variables. To this end, the Composite Responder Index in diffuse cutaneous SSc (CRISS) has been developed and is awaiting validation in a clinical trial.7 This index incorporates both mRSS and FVC, as well as the physician global assessment, the patient global assessment and the Health Assessment Questionnaire-Disability Index (HAQ-DI).
Current Therapeutics
Currently, no approved targeted therapies exist for the treatment of scleroderma. Treatment strategies have focused on traditional immunosuppressive therapies that have proven efficacy in other rheumatic conditions.
The two best studied medications in the treatment of SSc, and specifically SSc-ILD, have been cyclophosphamide and mycophenolate mofetil (MMF).
(click for larger image)
Figure 1: The pathogenesis of scleroderma. Vascular injury, immune system activation and tissue damage lead to the release of mediators that cause fibroblast activation, myofibroblast accumulation and ultimately the deposition of extracellular matrix. Note: ROS, reactive oxygen species; TLR, toll-like receptor, TGF-β, transforming growth factor beta; LPA, lysophosphatidic acid.
In the Scleroderma Lung Study I, a year of treatment with oral cyclophosphamide for SSc-ILD was found to be modestly better than placebo in stabilizing FVC at one year.8 However, in a follow-up study, this effect was reduced at two years, suggesting a transient benefit. In the recently completed Scleroderma Lung Study II, mycophenolate mofetil was compared to cyclophosphamide for the treatment of SSc-ILD.9 MMF was found to be non-inferior to cyclophosphamide with comparable improvements in FVC in both treatment groups at 24 months. Of note, the mRSS improved in both treatment arms with a trend favoring cyclophosphamide.
Based on the results of these two trials, first-line treatment for progressive SSc‑ILD will likely shift from cyclophosphamide to MMF given its more favorable side-effect profile. MMF has separately been evaluated for the treatment of scleroderma skin disease, albeit in small cohorts, and may have beneficial effects on lowering skin score, particularly in patients with early diffuse disease.10,11
With further advances in understanding the biology of the disease, stratifying patients, & determining comprehensive outcome measures for clinical trials, it is highly likely that effective therapies for scleroderma fibrosis will emerge in the next decade.
Other traditional disease-modifying anti-rheumatic drugs (DMARDs), including methotrexate, have been evaluated in scleroderma. A small randomized, controlled trial compared one year of methotrexate with placebo in patients with early (less than three years) diffuse scleroderma.12 Despite the small size of the study, there was a trend toward improvement in the mRSS in the methotrexate group. Based on this and another smaller study, the EULAR and European Scleroderma Trial and Research Group (EUSTAR) recommend methotrexate for skin disease in patients with early diffuse cutaneous SSc (dcSSc).13,14
Rituximab, a monoclonal antibody against CD20, used in the treatment of rheumatoid arthritis and ANCA-associated vasculitis, has come of interest in the treatment of SSc. A small nested case-control study demonstrated that patients treated with rituximab vs. matched controls had improvement in their mRSS, and prevented further decline of FVC in a small subset of patients with ILD.15 Based on this and other small studies, there is hope that rituximab may be beneficial in SSc, but larger studies are needed.16
Targeted Therapeutics
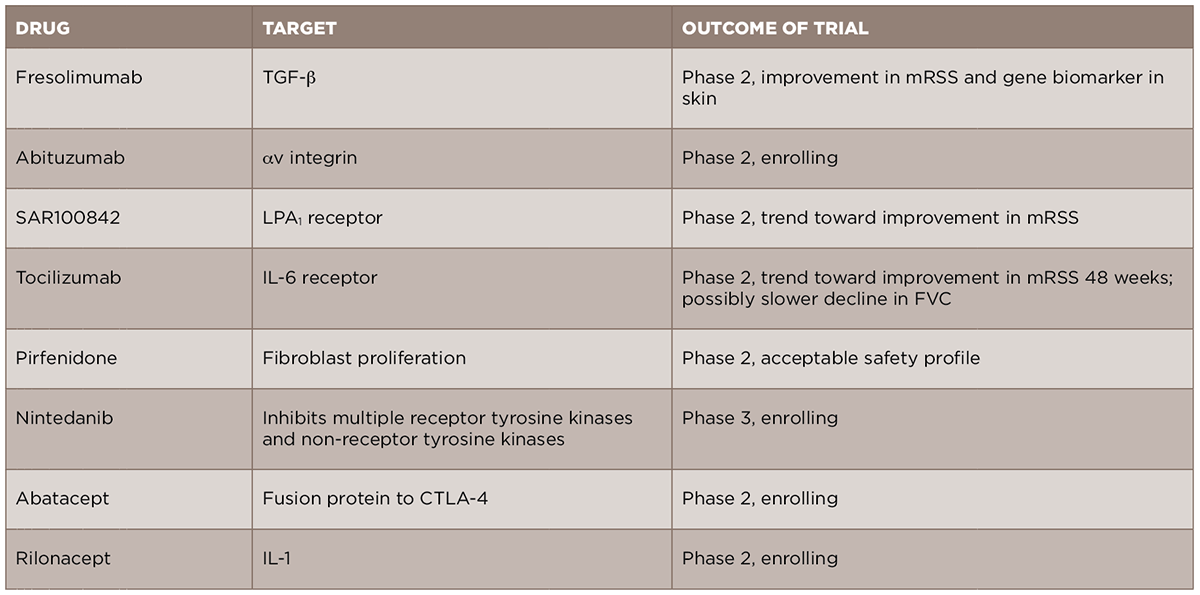
In light of the recent advances in the understanding of the pathogenesis of SSc, new anti-fibrotic treatments are currently in development and being studied for the treatment of the disease (see Table 2). We will review some of these new treatments by their molecular targets.
(click for larger image)
TABLE 2: New Targeted Therapeutics in Scleroderma Fibrosis
Given the central role of TGF-β in the development of scleroderma fibrosis, it is a natural target for therapy in SSc. A recent open-label trial of fresolimumab, a monoclonal antibody against TGF-β that neutralizes all isoforms of the molecule, compared two doses of the drug in patients with early (less than two years) dcSSc.17 The primary outcomes of this trial were change in mRSS and change in mRNA levels of two biomarkers measured in the skin, cartilage oligomeric protein (COMP) and thrombospondin-1 (THBS1), at 24 weeks. In both groups, there was a relatively rapid decrease in mRSS and in TGF-β regulated biomarker genes, in particular THBS1, suggesting that targeting TGF-β may be an effective treatment strategy. Of note, there were a number of cases of anemia in both groups, and this will need to be examined further in future, larger studies. Another drug targeting the TGF-β pathway, abituzumab, a monoclonal antibody against αv integrin, will be evaluated in an upcoming Phase 2 trial for SSc-ILD (ClinicalTrials.gov identifier: NCT02745145).
In addition to directly targeting TGF-β, a number of other important pathways can lead to the activation of fibroblasts and to the production of TGF-β. Based on the preclinical work of LPA and its receptor, LPA1, in dermal fibrosis, a Phase 2 trial of an LPA1 antagonist, SAR100842, was recently completed and showed an excellent safety profile and promising clinical efficacy.18 A Phase 3 trial of the LPA1 antagonist in scleroderma is currently being planned.
Given the role of endothelin-1 in fibrosis and the use of endothelin-1 receptor antagonists (ERAs) in the treatment of SSc-associated PH, there has been promise that these medications might additionally provide benefit in skin fibrosis. However, a recent cohort study of patients from the EUSTAR database compared patients treated with an ERA vs. those who had not, and failed to show a difference in the change in mRSS between the two groups over the study period.19 In addition, the Bosentan in Interstitial Lung Disease in Systemic Sclerosis-2 (BUILD‑2) study, which randomized patients with SSc‑ILD to bosentan or placebo, found no difference in the six-minute walk test or in pulmonary function tests at 12 months, further suggesting that this group of medications may not be useful for the fibrotic manifestations of SSc.20
Tocilizumab, a monoclonal antibody against the IL-6 receptor, is currently approved for use in rheumatoid arthritis and is under evaluation for several rheumatic diseases. A recent Phase 2 trial of tocilizumab in SSc was completed.21 Although the primary endpoint of a reduction in mRSS was not met at Week 24, there was a trend toward improvement in skin scores in the tocilizumab group compared to the placebo group at Week 48. Interestingly, there was also a trend toward a slower FVC decline, although the study was not initially designed to look at this endpoint. Based on these results, a Phase 3 trial is currently being planned.
A number of other new therapeutics are currently under evaluation for SSc fibrosis. Pirfenidone and nintedanib, two anti-fibrotic medications approved for the treatment of idiopathic pulmonary fibrosis (IPF), are currently being studied in SSc-ILD. A Phase 2 trial of pirfenidone in SSc-ILD (LOTUSS) was completed and showed acceptable tolerability, even with over 60% of patients concurrently taking MMF.22 A Phase 3 trial of nintedanib in SSc-ILD is currently ongoing and recruiting patients (ClinicalTrials.gov identifier: NCT02597933).
Abatacept, a CTLA4-IgG fusion protein approved for use in rheumatoid arthritis, is currently being studied in patients with early dcSSc (ASSET; ClinicalTrials.gov identifier: NCT02161406).
Rilonacept, an IL-1 inhibitor, is also being studied in scleroderma, using a four-gene biomarker of skin disease as the primary outcome in a short-duration trial (ClinicalTrials.gov identifier: NCT01538719).
Summary
There has been significant progress in understanding the pathogenesis of scleroderma and consequently in the development of novel targeted therapies for patients with this devastating disease. Until recently, treatment has focused on traditional immunosuppressive drugs and management of specific, organ-based complications with variable success. With further advances in understanding the biology of the disease, stratifying patients, and determining comprehensive outcome measures for clinical trials, it is highly likely that effective therapies for scleroderma fibrosis will emerge in the next decade.
 Sara R. Schoenfeld, MD, is a rheumatologist in the Division of Rheumatology at Massachusetts General Hospital and an instructor in medicine at Harvard Medical School in Boston.
Sara R. Schoenfeld, MD, is a rheumatologist in the Division of Rheumatology at Massachusetts General Hospital and an instructor in medicine at Harvard Medical School in Boston.
 Flavia V. Castelino, MD, is director of the Scleroderma Program in the Division of Rheumatology at Massachusetts General Hospital and assistant professor of medicine at Harvard Medical School in Boston.
Flavia V. Castelino, MD, is director of the Scleroderma Program in the Division of Rheumatology at Massachusetts General Hospital and assistant professor of medicine at Harvard Medical School in Boston.
References
- Mayes MD, Lacey JV Jr., Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug;48(8):2246–2255.
- Elhai M, Meune C, Avouac J, et al. Trends in mortality in patients with systemic sclerosis over 40 years: A systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2012 Jun;51(6):1017–1026.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013 Nov;65(11):2737–2747.
- Khan K, Xu S, Nihtyanova S, et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis. 2012 Jul;71(7):1235–1242.
- Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008 Jan;14(1):45–54.
- Castelino FV, Seiders J, Bain G, et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011 May;63(5):1405–1415.
- Khanna D, Berrocal VJ, Giannini EH, et al. The American College of Rheumatology provisional composite response index for clinical trials in early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2016 Feb;68(2):299–311.
- Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006 Jun 26;354(25):2655–2666.
- Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS-II): A randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016 Sep;4(9):708–719.
- Le EN, Wigley FM, Shah AA, et al. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann Rheum Dis. 2011 Jun;70(6):1104–1107.
- Mendoza FA, Nagle SJ, Lee JB, Jimenez SA. A prospective observational study of mycophenolate mofetil treatment in progressive diffuse cutaneous systemic sclerosis of recent onset. J Rheumatol. 2012 Jun;39(6):1241–1247.
- Pope JE, Bellamy N, Seibold JR, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum. 2001 Jun;44(6):1351–1358.
- van den Hoogen FH, Boerbooms AM, Swaak AJ, et al. Comparison of methotrexate with placebo in the treatment of systemic sclerosis: A 24 week randomized double-blind trial, followed by a 24 week observational trial. Br J Rheumatol. 1996 Apr;35(4):364–372.
- Kowal-Bielecka O, Landewe R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: A report from the EULAR scleroderma trials and research group (EUSTAR). Ann Rheum Dis. 2009 May;68(5):620–628.
- Jordan S, Distler JH, Maurer B, et al. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015 Jun;74(6):1188–1194.
- Daoussis D, Liossis SN, Tsamandas AC, et al. Experience with rituximab in scleroderma: Results from a 1-year, proof-of-principle study. Rheumatology (Oxford). 2010 Feb;49(2):271–280.
- Rice LM, Padilla CM, McLaughlin SR, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015 Jul 1;125(7):2795–2807.
- Khanna D, Denton CP, Jagerschmidt A, et al. SAR100842, an antagonist of lysophosphatidic acid receptor 1, as a potential treatment for patients with systemic sclerosis: Results from a phase 2a study. American College of Rheumatology Annual Meeting, Abstract #876. 2014.
- Jordan S, Distler J, Maurer B, et al. Effect of endothelin-1 receptor antagonists on skin fibrosis in scleroderma patients from the EUSTAR database. JSRD. 2016;1(2):220–225.
- Seibold JR, Denton CP, Furst DE, et al. Randomized, prospective, placebo-controlled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010 Jul;62(7):2101–2108. [Erratum in Arthritis Rheum. 2010 Oct;62(10):3005.]
- Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet. 2016 Jun 25;387(10038):2630–2640.
- Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: The LOTUSS trial. J Rheumatol. 2016 Jul 1. pii: jrheum.151322.
