Editor’s Note: The nomenclature for Wegener’s recently changed from Wegener’s granulomatosis to granulomatosis with polyangiitis (Wegener’s). Beginning with the June 2011 publication of The Rheumatologist, the new nomenclature will be used. See “Medical Societies Ask, What’s in a Name?“ on p. 48 for more on the evolution of disease nomenclature.
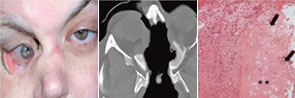
Wegener’s granulomatosis (WG) is per definitionem a granulomatous disorder involving the respiratory tract and is usually associated with vasculitis, affecting small to medium-sized vessels and the production of antibodies to neutrophil cytoplasmic antigens (ANCA) directed to the antigen proteinase 3 (PR3).1 Although vasculitis may be its classical feature, WG also may occur as a persistent granulomatous inflammatory process without apparent vasculitis.2 While there is no clear definition of granulomatous disease in WG, this pathological process relates to the formation of masses (often round in appearance on radiograph) or the histological picture of granuloma that is frequently detected within these masses. We know about the vasculitic side of WG’s coin; much less is known about the granulomatous side of WG, however. In this article, we will focus on recent clinical and immunopathological findings attributed to the so-far enigmatic granulomatous process and the probable link to the development of PR3-ANCA and vasculitis.3
Categorizing Disease Stages
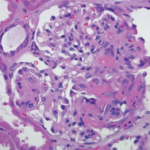
WG is commonly considered to be part of the spectrum of disorders characterized by vasculitic lesions throughout the body without a known cause but associated with autoantibodies against PR3-ANCA.4 Therefore, WG is included into the group of diseases known as primary systemic vasculitides and grouped together with others (e.g., microscopic polyangiitis, Churg-Strauss syndrome) also strongly associated with ANCA and “pauci-immune” glomerulonephritis in situ.2 There is strong evidence for the pathogenic role of antibodies against myeloperoxidase (MPO-ANCA) in inducing vasculitis, and this mechanism may also play a role in PR3-ANCA associated vasculitis such as WG.5 However, the clinical and pathological hallmark of WG is not only vasculitis but the coexistence of vasculitis and granuloma, both of which differentiate WG from microscopic polyangiitis. The presence of two distinct pathological processes needs to be considered in categorizing disease stages in WG.
right panel, Arthritis Rheum. 2008;58:834.
Reprinted with permission.
Currently, there are two approaches to categorize disease stages in ANCA-associated vasculitis (AAV). In the United States, it is common to differentiate a limited form of WG in which patients suffer from non–life-threatening vasculitis manifestations from a severe form with life-threatening vasculitic manifestations, such as glomerulonephritis.6 In contrast, the European approach separates a disease stage with granulomatous manifestations of the upper and lower respiratory tract (and with no clinical signs of vasculitis) from systemic disease stages with granulomatous and vasculitic manifestations (early systemic, non–life-threatening; generalized, organ-threatening; and severe with organ failure).7,8