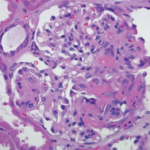
Regarding localized disease manifestation, mucosal ulceration leading to bloody rhinitis is seen by nasal endoscopy during the beginning of the disease; later, septum infiltration and perforation may occur as well as the destruction of the conchae. Granulomatous masses may be detected by MRI imaging in the sinuses and can be accompanied by bone destruction of the adjacent sinus, ethmoidal cysts, and orbita, leading to proptosis and compression of the optic nerve.13 In addition, per vias naturales the masses can obstruct the lacrimal duct or the subglottic region and, less frequently, bronchi, which may cause subsequent poststenotic pneumonia. Via the ethmoidal cysts, the process rarely invades the intracranial space and brain. In the lung, the masses usually appear as round nodules with a tendency to cavitate (see Figure 1, above).
Damage Induced by Granulomatous Inflammation
Although advances in the treatment of WG have resulted in a dramatic reduction in disease-related mortality, patients still experience relapses due to the vasculitis and the granulomatous process and side effects from treatment toxicity.11 Using damage scores to assess the total burden of damage experienced by the patient, it can be demonstrated that, besides the damage associated with malignancy, tissue ischemia, and organ failure, items of damage obviously associated with the granulomatous process were frequently scored and rated as medium to severe damage items.11 Examples of damage related to granulomas include proptosis, pseudotumor, nasal bridge collapse, subglottic/ large airway obstruction, orbital wall destruction, diplopia, and optic nerve atrophy. These findings show that the so-called granuloma include space-occupying lesions and tissue-destructive events.
right panel, Rheumatology. 2008;47:1111-1113.
Reprinted with permission.
Localized Granulomatosis as a Persistent Variant of WG
Localized WG may occur not only as a short-term disease stage before generalized disease develops. Recently, this form of disease also has been identified by two European centers as a persistent variant.9,10 In our study, persistent localized WG was observed in 5% of patients in a cohort of 1,024 patients with biopsy-compatible WG followed from 1989 to 2009.9 Only around 50% of the patients were ANCA positive. Localized disease manifestations were characterized by destructive and space-occupying lesions and associated with a high rate of organ damage. Furthermore, nearly all patients with granulomatous involvement (89% over the whole course of follow-up) required medium or highly potent immunosuppression such as methotrexate or cyclophosphamide for remission induction. Cotrimoxazole, which was used in half of the patients as initial treatment, was shown to be insufficient to control disease activity in 72%. In spite of close follow-up and step-up of immunosuppression in cases of persistent or refractory disease activity, 66% of patients acquired at least some kind of organ damage (i.e., saddle nose deformity, septal perforation, bony destruction of sinus and orbital walls).