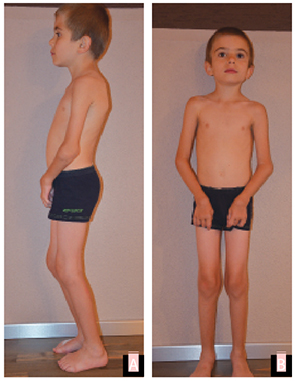
On examination, he presented with significant limitation of motion of fingers II through V of both hands with thickening of the flexor tendon sheaths. He also had swelling of the ankles, knees, wrists and elbows, with significant pain and limitation of motion. Ultrasound at the time of presentation showed only mild effusions in the elbows, with no synovial thickening, but tenosynovitis of the flexor tendon sheaths. No subcutaneous nodules were noted.
Laboratory investigations showed normal CBC, negative CRP and ESR, and normal values for renal and liver function tests. Serum immunoglobulin levels for IgG, IgA and IgM were within normal range. Rheumatoid factor and antinuclear antibodies were not present, and HLA-B27 was negative.
He was tentatively diagnosed with juvenile idiopathic arthritis (with a subtype according to ILAR criteria of rheumatoid factor-negative polyarthritis) and was treated with indomethacin (2.7 mg/kg/day, divided three times per day), subcutaneous methotrexate (12.5 mg/m2 BSA/week) and several courses of methylprednisolone, which led to only temporary improvement.