
Anton Khrupin / Shutterstock.com
A 58-year-old African American woman with a past medical history of hypertension (HTN), hyperlipidemia, severe gastroesophageal reflux disease (GERD) and limited cutaneous systemic sclerosis (lcSSc) presented to the emergency department with shortness of breath (SOB) and progressive bilateral lower extremity swelling for three weeks. She denied any chest pain, but endorsed generalized fatigue and dyspnea with activities of daily living. She denied orthopnea, but stated she always sleeps upright secondary to severe GERD. On review of systems, she endorsed occasional palpitations without presyncopal symptoms and a dry cough. She denied any caffeine intake, stimulant use, alcohol use or illicit drug use.
Her medications included a daily low-dose aspirin, triamterene-hydrochlorothiazide, pravastatin, omeprazole, amlodipine for Raynaud’s symptoms and infrequent use of ibuprofen as needed for pain or headaches.
Vital signs at triage showed elevated blood pressure of 160/114 with tachycardia at 152 beats per minute (bpm), mild tachypnea with a respiratory rate of 22 breaths per minute and 98% oxygen saturation on room air. After she was given furosemide and metoprolol intravenously, her blood pressure improved to 124/80 with continued tachycardia at 124 bpm. Physical exam revealed respiratory distress, elevated jugular venous pressure, bibasilar crackles and skin tightening on the bilateral dorsal aspects of her hands and distal forearms with digital necrosis of her right third digit (see Figure 1). No murmurs were auscultated, and her tachycardia had a regular rhythm.
Laboratory evaluation revealed an elevated brain-type natriuretic peptide, acute kidney injury, a bland urinalysis and an elevated troponin. Her lcSSc serologies were significant for positive anti-nuclear antibody 1:160 speckled pattern, positive anti-Ro antibody and negative anti-topoisomerase antibody. Her admission electrocardiogram (ECG) was interpreted as demonstrating a narrow complex tachycardia at 130 bpm (see Figure 2), most consistent with sinus tachycardia.
A CT of her chest, pulmonary embolism protocol, showed bilateral pleural effusions without evidence of parenchymal abnormalities consistent with interstitial lung disease (see Figure 3). An echocardiogram showed a left ventricular ejection fraction of 33%, concentric left ventricular hypertrophy, and an estimated systolic pulmonary artery pressure of 80 mmHg. Cardiac catheterization showed normal coronary arteries, normal pulmonary artery capillary wedge pressure and a pulmonary artery pressure mean of 38 mmHg, indicating mild pulmonary hypertension.
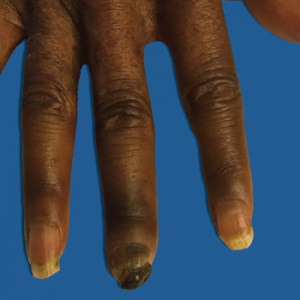
Figure 1: Right Third Digit Necrosis
Patient has mild edema and significant loss of nail, as well as clinical evidence of acroosteolysis of her third digit.
Upon discharge, her diagnoses included hypertensive emergency, acute chronic heart failure with reduced ejection fraction, acute renal failure without evidence of scleroderma renal crisis and mild pulmonary hypertension. Given her dangerously low left ventricular ejection fraction, which can be associated with sudden cardiac death, she was discharged with an external defibrillator vest.
