Discussion
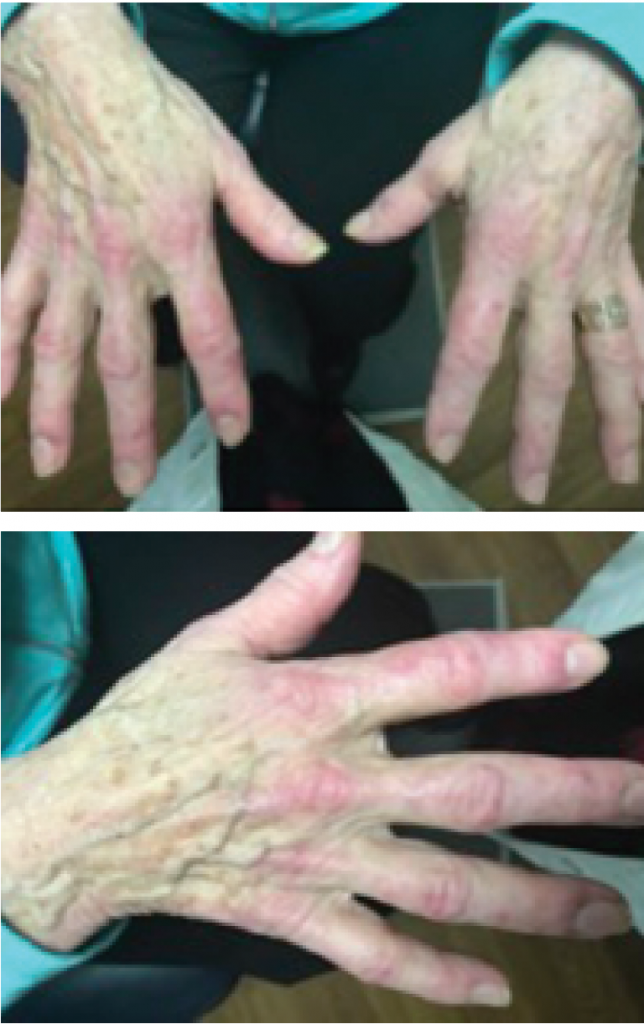
Figures 1 & 2: A hallmark skin manifestation of dermatomyositis includes Gottron’s papules, lesions consisting of erythematous to violaceous papules and plaques found over the metacarpophalangeal and interphalangeal joints.
Carcinoid syndrome is a paraneoplastic constellation of signs and symptoms mediated by the amplification of humoral factors by well-differentiated neuroendocrine tumors (NETs). These tumors, usually located in the digestive tract and lungs, secrete various polypeptides, biogenic amines and prostaglandins, such as serotonin and kallikrein. The annual incidence of NETs is anywhere from 2.5–5 per 100,000; the prevalence is estimated to be around 35 per 100,000. Postmortem exams have suggested asymptomatic NETs may be more common than reported, and reports of incidental discoveries of non-functioning tumors exsist. Symptoms associated with NETs include cutaneous flushing, secretory diarrhea, bronchospasms and cardiac valvular lesions. Typically, a NET is suspected when these symptoms are unexplained.1
Diagnosis of NETs involves laboratory testing, which includes serum chromogranin concentration, blood serotonin concentration, plasma 5-HIAA concentration and urinary excretion of serotonin and 5-HIAA. Various imaging modalities, such as computed tomography (CT) scan and magnetic resonance imaging, are used to isolate the NET. Indium-111 pentetreotide detects somatostatin receptors, which are expressed in high levels in NETs and are also useful to detect these tumors.