Clinically, the dermatologist noted a heliotrope eruption, Gottron’s papules, a shawl sign and a V sign. A skin biopsy was consistent with dermatomyositis. She began treatment with prednisone and methotrexate, and was referred to our office for further management.
Although it’s generally accepted dermatomyositis patients should be screened for cancer, no consensus exists as to what screening should entail.
The patient’s laboratory tests did not demonstrate an antinuclear antibody, anti-double stranded DNA antibody or rheumatoid factor. Muscle enzymes, including creatine kinase and aldolase, were within normal limits, although mild proximal muscle weakness existed on examination. Noteably, her chromogranin A was elevated at 70 nmol/L (upper limit of normal [ULN]: 15 nmol/L), and her 24-hour urine 5-HIAA was 9.2 mg/24h (ULN 5.99 mg/24h).
Due to the persistent rash, the patient’s methotrexate dose was increased from 15 to 20 mg weekly. She was also instructed to remain on prednisone. Initial labs, including a myositis antibody panel, demonstrated the following:
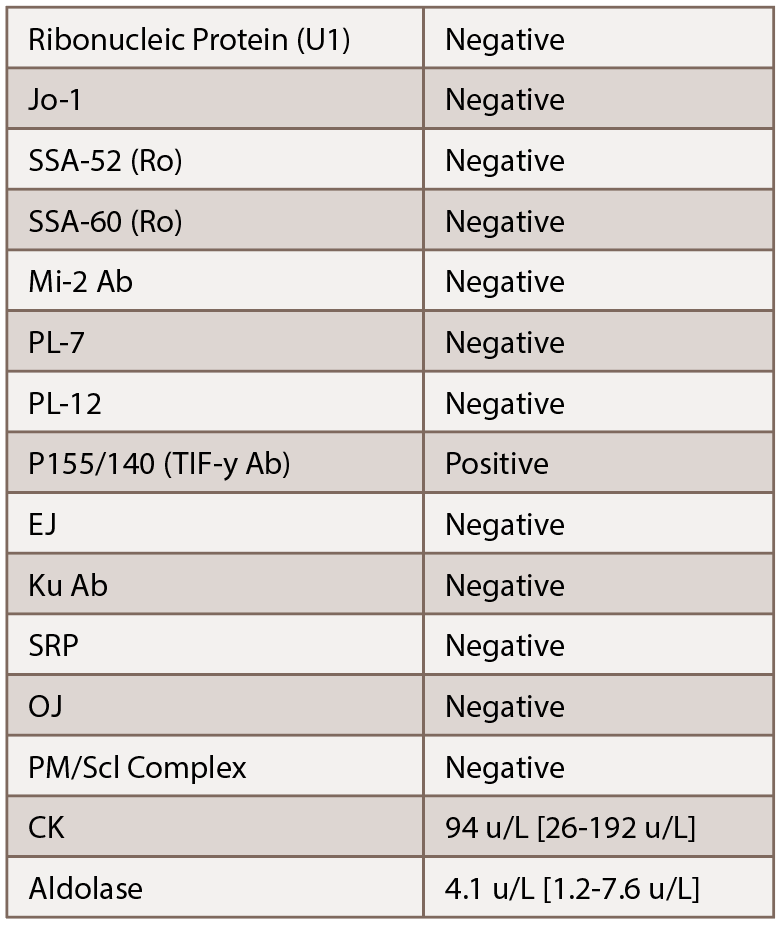
Given the elevated p155/140 antibody, we were concerned about a cancer-associated dermatomyositis. Additionally, given her lack of significant muscle involvement with normal enzymes, we suspected an amyopathic variant. With her history of an undifferentiated neuroendocrine tumor, recommendations were made for her to follow up with her oncologist for further evaluation. Subsequent lab tests demonstrated her chromogranin A increased to 82 nmol/L.
Despite immunosuppression, the patient’s rash persisted. Hydroxychloroquine was added as a steroid-sparing agent for her skin manifestations, and eventually, mycophenolate mofetil was added as well. The patient’s oncologist placed her on octreotide therapy to treat her carcinoid tumor. With the addition of octreotide, her rash resolved. However, her symptoms returned when the therapy was stopped.
The rash recurred mainly on the patient’s upper chest. It was initially intermittent, but gradually became persistent and was associated with flushing and pruritis. Her heliotrope eruptions and Gottron’s papules ultimately resolved. At follow-up visits, it was determined her recurrent rash was due to carcinoid rather than her dermatomyosits.
Discussion
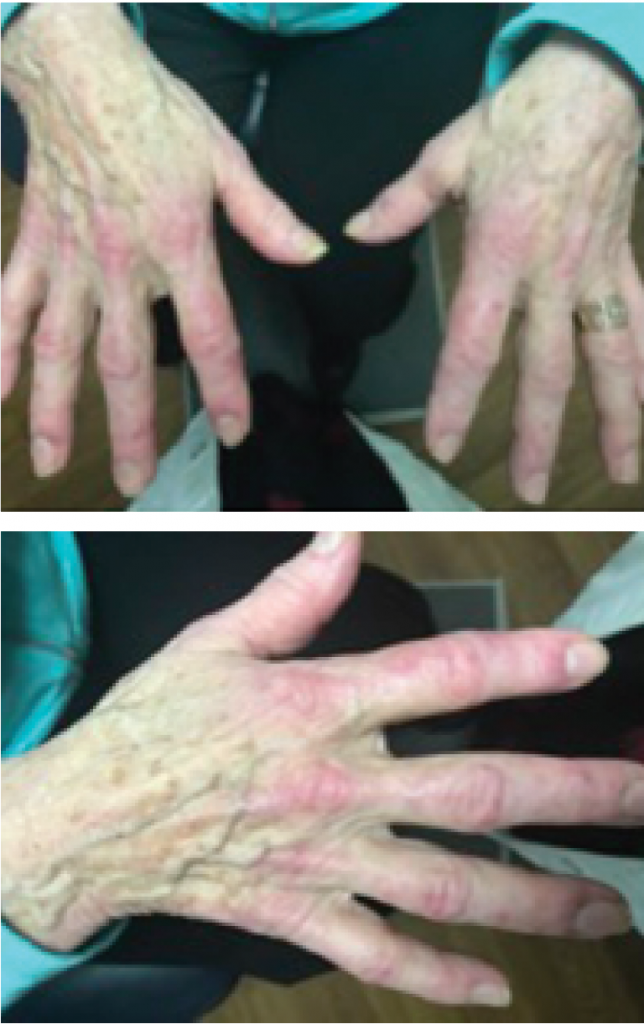
Figures 1 & 2: A hallmark skin manifestation of dermatomyositis includes Gottron’s papules, lesions consisting of erythematous to violaceous papules and plaques found over the metacarpophalangeal and interphalangeal joints.
Carcinoid syndrome is a paraneoplastic constellation of signs and symptoms mediated by the amplification of humoral factors by well-differentiated neuroendocrine tumors (NETs). These tumors, usually located in the digestive tract and lungs, secrete various polypeptides, biogenic amines and prostaglandins, such as serotonin and kallikrein. The annual incidence of NETs is anywhere from 2.5–5 per 100,000; the prevalence is estimated to be around 35 per 100,000. Postmortem exams have suggested asymptomatic NETs may be more common than reported, and reports of incidental discoveries of non-functioning tumors exsist. Symptoms associated with NETs include cutaneous flushing, secretory diarrhea, bronchospasms and cardiac valvular lesions. Typically, a NET is suspected when these symptoms are unexplained.1
Diagnosis of NETs involves laboratory testing, which includes serum chromogranin concentration, blood serotonin concentration, plasma 5-HIAA concentration and urinary excretion of serotonin and 5-HIAA. Various imaging modalities, such as computed tomography (CT) scan and magnetic resonance imaging, are used to isolate the NET. Indium-111 pentetreotide detects somatostatin receptors, which are expressed in high levels in NETs and are also useful to detect these tumors.
Traditionally, NETs have been classified by location, with hundreds of anatomic locations possible. To simplify classification, the World Health Organization has introduced a three-tier grading system, emphasizing tumor grade and not anatomic origin. NETs are staged depending on location, invasion and size.
Treatment and management for NETs depend on staging, grade and functionality of the tumor. Often, an important role for surgical intervention exsists to some degree. When possible, complete surgical excision of a NET is performed. After resection, patients should continue to be screened with intermittent serum and urine tests, as well as imaging.
Other treatments are aimed at palliation. For non-functioning, low-grade NETs, close observation can be offered. Symptomatic relief involves management with somatostatin analogs, such as octreotide. Anti-diarrheal medications may also be used. Cytotoxic chemotherapy is used for high-grade NETs. And radionuclide therapy has been shown to be effective for more aggressive NETs (i.e., if the tumor has high uptake of somatostatin). Radiofrequency ablation and cryoablation are also used to treat NETs. Gene therapies are currently in Phase 1 trials.2
NETs are very rarely linked to dermatomyositis. In literature, only a handful of case reports link NETSs to dermatomyositis, with the majority involving NETs in the liver or lungs. In most cases, treatment of the NET resolved the rheumatologic issue.
Dermatomyositis
Dermatomyositis is characterized by proximal, symmetric muscle weakness and skin eruption. Comprehensive epidemiologic data are lacking, because most studies are limited due to variations in age, gender and region. Moreover, most studies rely on physician billing and hospitalization databases, which may not capture all diagnoses.3 Nevertheless, Bendewald et al., using a 32-year, population-based retrospective study from Olmsted County, Minn., estimated that after age and sex adjustment, the incidence of dermatomyositis was about 1 per 100,000 people per year.4 The prevalence was around 20 cases per 100,000 people. Generally, dermatomyositis has a bimodal distribution that occurs in two peaks: one in the early teenage years and the second when the patient is 45–64 years old.
Five major criteria established by Bohan et al. in 1975 may be used to define dermatomyositis.5 These criteria include, 1) symmetrical weakness of the limb-girdle muscles and anterior neck flexors progressing from weeks to months with or without dysphagia or respiratory muscle involvemen; 2) muscle-biopsy evidence of necrosis of type I and II fibers, phagocytosis, regeneration with basophilia, large vesicular sarcolemmal nuclei and prominent nucleoli, atrophy in a peri-fascicular distribution, variation in fiber size, and an inflammatory exudate; 3) elevation in serum of skeletal-muscle enzymes, such as creatine phosphokinase and/or aldolase; 4) electromyographic triad of short, small, polyphasic motor units, fibrillations, positive sharp waves and insertional irritability, and bizarre, high-frequency repetitive discharges; and 5) dermatologic features, including heliotropic rash with periorbital edema and/or a scaly, erythematous dermatitis over the dorsum of the hands (e.g., Gottron’s papules) and involvement of knees, elbows, malleoli, face, neck or upper torso.
Three or four criteria, with the rash, can be considered definite for dermatomyositis. Two criteria with rash is considered probable, while one criterion plus rash can be considered possible.
It’s thought that 50–70% of dermatomyositis patients will have myositis-specific antibodies against nuclear or cytoplasmic antigens.6 Tumor-associated autoimmunity can also be directed against mutated forms of self-antigens or unrelated antigens associated with paraneoplastic syndromes. Examples include:
- Antibodies to Yo/CDR2 antigen in paraneoplastic cerebellar degeneration, which are associated with breast and ovarian cancers;
- Antibodies against Ri/NOVA (neuro-oncological ventral antigen) in paraneoplastic opsoclonus-myoclonus ataxia in those with small cell lung, breast and ovarian cancers; and
- Antibodies against TIF-1-gamma (p155/140 antibody), which are hypothesized to be overexpressed in certain malignancies.
Why Should We Care?
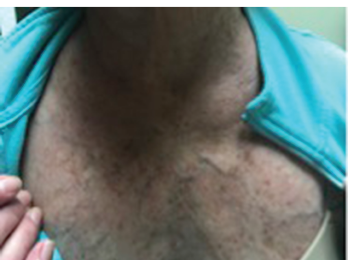
Figure 3: The flushing rash of carcinoid can mimic the rash of dermatomyositis, particularly the V sign, which refers to macular erythema over the anterior neck and upper chest.
P155/140 antibodies, which target transcriptional nuclear factor TIF-1-gamma, were first described by Targoff et al.7 It is of interest because it could potentially predict which dermatomyosits patients have an underlying malignancy. Prior to this, it was noted the presence of other myositis-specific antibodies decreased the likelihood of concurrent malignancy. Kaji, et al. further confirmed this, but their sample sizes were small.8 Later on, Chinoy et al. analyzed a large cohort of inflammatory myopathy patients and found anti-155/140 is dermatomyositis specific, and out of the 16 cancer-associated myositis patients, eight were positive for this antibody, implying 50% sensitivity and 100% specificity.9
Screening for malignancies in dermatomyositis patients
Although it’s generally accepted dermatomyositis patients should be screened for cancer, no consensus exists as to what screening should entail. Experts have suggested blind testing is not helpful, while others say CT scans and gastrointestinal endoscopy may play a useful role.
In the largest study to date, two large dermatology cohorts were used to identify 400 patients with dermatomyositis.10 In this study, 15.8% of patients had cancer, 72% occurring within five years of dermatomyositis-symptom onset, suggesting a temporal component. In descending order, the most frequent types of cancer were breast, hematologic and colorectal. The vast majority of the cancers (42%) were diagnosed after the dermatomyositis diagnosis and were associated with suspicious signs, such as unintentional weight loss, melena or persistent abdominal pain. It seems reasonable to screen all patients with dermatomyositis for malignancy using age-appropriate guidelines.
The difference between carcinoid & dermatomyositis skin manifestations
Several hallmark skin manifestations of dermatomyositis exist. Gottron’s papules are lesions consisting of erythematous to violaceous papules and plaques found over the extensor surfaces of the metacarpophalangeal and interphalangeal joints. These lesions develop scaling that can resolve with dyspigmentation, atrophy and scarring. Gottron’s sign refers to erythematous macules and patches that develop over the extensor surfaces of the elbows and knees. The V sign refers to macular erythema over the anterior neck and upper chest, whereas the shawl sign occurs on the upper back, shoulders and posterior neck. Finally, a heliotrope rash is described as violaceous erythema of the upper eyelids with edema and telangiectasias.4
The rash of carcinoid tumors is different in several ways. First, carcinoid tumors secrete biologically active substances, such as serotonin (5-HT), substance P, histamine, catecholamines and prostaglandins, that escape inactivation by hepatocytes and induce flushing. Midgut tumors produce rashes that are generally rapid, cyanotic and last for less than a minute. These are associated with a mild burning sensation. Foregut tumors produce wheals with pruritus that are reddish-brown and occur over the entire body. Location, lack of scale and pattern distinguish the rash of carcinoid from that of dermatomyositis. Looking for the characteristic symptoms associated with carcinoid can also be very helpful.
Conclusion
This was an interesting case of cancer-associated dermatomyositis associated with an undifferentiated neuroendocrine carcinoid tumor with bilateral ovarian and bowel involvement. The flushing rash of carcinoid can mimic the rash of dermatomyositis, particularly the V sign.
With the identification of new autoantibodies in the inflammatory myopathies, more associations will be made. We must continue to try to determine which of these novel antibodies are associated with cancer-associated dermatomyositis. We must also continue to recognize the variety of malignancies associated with this condition. Finally, more research into how to screen these patients for cancer is needed.
Osman Bhatty, MD, is a chief resident at Montefiore Medical Center Wakefield Campus, Bronx, N.Y.
Rouhin Sen, MD, is a chief resident at Creighton University Medical Center, Omaha, Neb.
Joseph Nahas, MD, is an academic rheumatologist and internal medicine program director at Creighton University Medical Center, Omaha, Neb.
References
- Dasari A, Shen C, Halperin D, et al. Trends in the incidence, prevalence and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017 Oct 1;3(10):1335–1342.
- Young K, Iyer R, Morganstein D, et al. Pancreatic neuroendocrine tumors: A review. Future Oncol. 2015;11(5):853–864.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012 Sep;57(5):375–381.
- Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010 Jan;146(1):26–30.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975 Feb 13;292(7):344–347.
- Madan V, Chinoy H, Griffiths CE, Cooper RG. Defining cancer risk in dermatomyositis. Part II. Assessing diagnostic usefulness of myositis serology. Clin Exp Dermatol. 2009 Jul;34(5):561–565.
- Targoff IN, Trieu EP, Sontheimer RD. Autoantibodies to 155 kD and Se antigens in patients with clinically amyopathic dermatomyositis [abstract]. Arthritis Rheum. 2000;43(suppl 9):S194.
- Kaji K, Fujimoto M, Hasegawa M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: An association with malignancy. Rheumatology (Oxford). 2007 Jan;46(1):25–28.
- Chinoy H, Fertig N, Oddis CV, et al. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. 2007 Oct;66(10):1345–1349.
- Leatham H, Schadt C, Chisolm S, et al. Evidence supports blind screening for internal malignancy in dermatomyositis: Data from 2 large U.S. dermatology cohorts. Medicine (Baltimore). 2018 Jan;97(2):e9639.