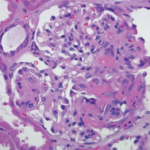
Over the course of the next year, the patient has experienced complete resolution of virtually all of his symptoms, has been successfully tapered off of prednisone without any recurrence of symptoms and is tolerating methotrexate without any major adverse reactions. He continues to follow up with rheumatology regularly.
Discussion
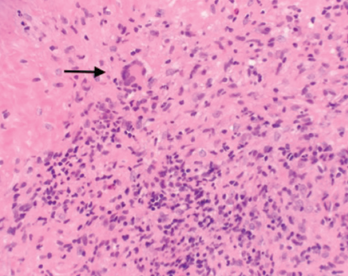
Figure 2. A nasal biopsy shows giant multinuclear cells (black arrow).
GPA is a complex, multisystem disease that is often difficult to diagnose, especially at the initial presentation. Early diagnosis is critical to halt the disease process and progression. Patients with GPA diagnosed early in the disease process, especially in the ambulatory setting, have had favorable outcomes compared with patients enrolled in randomized controlled trials who are usually referred to tertiary centers and present with advanced disease.4 This can certainly be attributed to the delay in diagnosis and or misinterpretation of initial presenting symptoms.