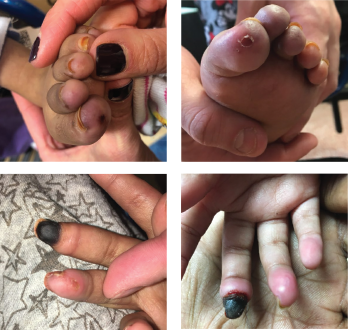
Figures 1A-D: A 5-year-old girl presented with erythema and ulcerations of the finger- and toetips, which had progressed to ischemia of the right, third fingertip.
Janus kinase 1 and 2 inhibitors (jakinibs) have been approved for the treatment of rheumatoid arthritis, psoriatic arthritis and, most recently, juvenile idiopathic arthritis. They have also shown promise in the treatment of interferon (IFN) mediated diseases. The Janus kinase and signal transducer and activator of transcription (JAK/STAT) pathway is the principal signaling pathway for IFN receptors, which are upregulated in IFN-mediated diseases, such as Aicardi-Goutières syndrome.
Here, we present a case of a child who presented with a necrotic digit and was found to have Aicardi-Goutières syndrome, which had previously been misdiagnosed as metachromatic leukodystrophy. She would potentially have benefited from treatment with a jakinib, but was unable to obtain insurance approval for this medication.