Another feature of Aicardi-Goutières syndrome, observed in about 40% of cases and more commonly seen in those with the SAMHD1 mutation, is perniosis or chilblains, as seen in our patient.5,8 This is one of the most distinctive extra-neurological manifestations of Aicardi-Goutières syndrome and highlights the overlap of Aicardi-Goutières syndrome with some of the pathophysiology seen in systemic lupus erythematosus, such as disturbance of IFNα metabolism.1,3
Failure of primer repair exonucleases has been presumed to result in intracellular DNA/RNA, which triggers an inappropriate immune response and overproduction of IFNα.3,8 This overproduction can result in an IFN-induced vasculitis, manifesting as skin lesions and leukodystrophy, which can be seen in Aicardi-Goutières syndrome.
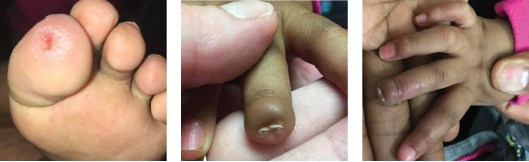
Figure 2: The patient’s erythema and ulcerations improved, and her ischemic digit auto-amputated.
With this in mind, the team revisited the patient’s diagnosis of metachromatic leukodystrophy. Further review of her history revealed that she had a normal birth and was initially developing normally. However, when she did not crawl or stand up when expected, she was referred to a neurologist and brain imaging was performed, which resulted in the diagnosis of metachromatic leukodystrophy.
Magnetic resonance imaging (MRI) of the brain was repeated and showed periventricular, white matter abnormalities consistent with metachromatic leukodystrophy, but also consistent with Aicardi-Goutières syndrome.
The patient’s arylsulfatase A level was found to be within normal ranges, providing strong evidence against a diagnosis of metachromatic leukodystrophy. Genetic testing revealed a homozygous mutation in the SAMHD1 gene—one of the gene mutations identified in patients with Aicardi-Goutières syndrome.
Upon further review of the family history, it was discovered the parents were second cousins. Genetic testing of her siblings found four of six had heterozygous mutations, but were otherwise healthy. (Note: Heterozygous individuals can also have clinical manifestations of this disease, so her siblings could now be monitored and receive interventions as appropriate.)
Our patient was treated with systemic glucocorticoids, azathioprine, aspirin, amlodipine and topical treatments for the perniosis. Her erythema and ulcerations improved (see Figure 2A). Eventually, her fingertip auto-amputated (see Figures 2B–C), and she did not have initial recurrence of her perniosis.
The family’s language barrier, low health literacy and obligations of caring for six other children made treatment adherence difficult, especially when it came to following a steroid taper. Given our patient’s risk of eventual relapse and in an effort to simplify her medical regimen while improving her overall outcome, we attempted to start a JAK 1/2 inhibitor (jakinib), given the promising data surrounding this class of medication for the treatment of interferonopathies.2,4,6,7
In one case study of an adult patient who also had perniosis in the setting of Aicardi-Goutières syndrome, the patient achieved complete remission with baricitinib.4 A recent study published in the New England Journal of Medicine evaluated 35 patients with genetically confirmed Aicardi-Goutières syndrome, and compared their interferon signaling gene expression score, symptoms associated with their disease and symptom development from before the patients started treatment with baricitinib with scores and symptoms found during treatment. The researchers found that interferon signaling gene expression scores decreased during treatment, as did reported symptoms by parents during treatment. Perhaps most important, many more children gained developmental milestones during treatment than before, and the number of milestones gained seemed to correlate with a higher dose of baricitinib.7
Unfortunately, our patient’s insurance repeatedly denied baricitinib or other jakinibs, despite our strong petitioning and legal hearing. At the time of this report, she was lost to follow-up. The stress from having to involve the legal system in attempts to get this medication approved may have contributed to this loss.
Discussion
This case reminds us to consider interferon-mediated diseases when presented with patients with vasculopathies and neurologic dysfunction. The success of correcting this patient’s diagnosis came from the team’s ability to take a step back and question what was already felt to be known—an important part of what rheumatologists do every day. Given the rarity of these diseases, patients with interferon-mediated diseases may often be misdiagnosed. Having a high index of suspicion when we see the combination of vasculopathy and neurologic dysfunction can avoid a delay in treatment, which can potentially prevent permanent, neurologically devastating effects of these diseases.
This case also considers the exciting promise of jakinibs in the treatment of an increasing number of autoimmune diseases, as well as the ongoing need for more research into this class of medications to explore their benefits, elucidate optimal doses and characterize side effects, among other factors.
Lastly, this case also serves as an all too common example of how social determinants of health can magnify disease burden and also serves as a reminder that addressing access to care, coverage of new therapies and culturally appropriate care are just as important as investing in exciting new pharmaceuticals. We undo much of the promise of new treatment options when the patients who could most benefit from them cannot receive them.