Treatment with anticoagulation and immunosuppression was initiated. She received high-dose prednisone (1 mg/kg) and was started on azathioprine (1 mg/kg; 50 mg twice daily), with resolution of her headache within the first two weeks of treatment. Prednisone was gradually tapered over 12 months without signs or symptoms concerning for relapse. For anticoagulation, she initially received unfractionated heparin and was then transitioned to warfarin to complete a total of eight months of treatment. The duration of anticoagulation was determined on the basis of resolution of the thrombi on subsequent imaging at three and six months.
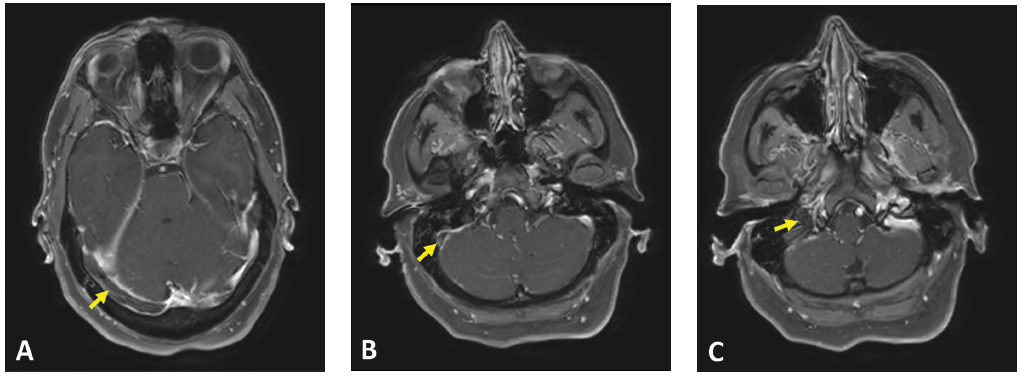
(click for larger image) Figure 1: Axial T1 contrast MR image showing non-enhancement within the (A) right transverse sinus, (B) right sigmoid sinus and (C) right internal jugular vein. Associated dural enhancement is visualized around the transverse sinus (A) and sigmoid sinus (B).
Behçet Disease
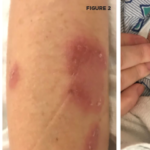
Behçet’s disease was first described by Hulusi Behçet, a Turkish dermatologist, in 1937 and was defined as a syndrome of recurrent oral aphthous ulcers, genital ulcers and uveitis.1 It’s classified as a variable vessel vasculitis (can affect vessels of any size and type), affecting both arteries and veins.2 The definition of Behçet’s disease has been refined over the years and the most widely used diagnostic criteria today are based on the International Study Group Diagnostic Criteria published in 1990. These include recurrent oral ulcerations of at least three incidents in a 12-month period, in addition to two other manifestations of recurrent genital ulcers, eye lesions, skin lesions or a positive pathergy test.1,3
In 2014, classification criteria were revised by the International Criteria for Behçet’s Disease (ICBD), which increased their sensitivity by including neurological and vascular manifestations in addition to oral and genital ulcerations. A score of 4 or more points is estimated to have 93.9% sensitivity and 92.1% specificity.4