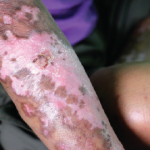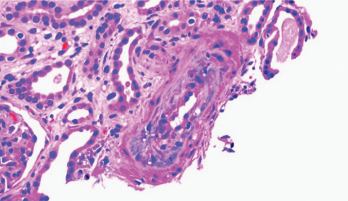
Figure 1. Thickening of the media layer of the vessel with narrowing of the arterial lumen is shown.
Scleroderma renal crisis (SRC) is a life-threatening complication of systemic sclerosis. SRC occurs in 2–15% of patients with diffuse sclerosis and usually within the first five years from the time of diagnosis.
Risk factors for SRC include, but are not limited to, early diagnosis, corticosteroid or cyclosporine use, and the presence of anti-RNA polymerase III antibodies. Usually, presenting symptoms include acute onset of hypertension and rapid deterioration of renal function.1 About 10% of patients present with normal blood pressure, making diagnosis more challenging and often delaying treatment. The prognosis is poor, with a five-year survival rate of less than 60%.1
The patient was also evaluated by a dermatologist. Two skin biopsies were obtained. The biopsy of the inner thigh was consistent with neutrophilic urticaria, and the biopsy from the forearm was consistent with morphea profunda. The patient was started on 20 mg of methotrexate weekly by the dermatologist, but developed nausea, vomiting and abdominal pain, so methotrexate was discontinued and high-dose prednisone (60 mg/day) was initiated.
At the time of admission
One month after starting the prednisone, the patient presented to the emergency department with complaints of severe abdominal pain, nausea, vomiting, jaundice, decreased appetite, unintentional 20 lb. weight loss, chills and fevers. On physical exam, the patient was tachycardic with a heart rate of 115. She was lethargic and pale, with scleral icterus. She also had weakness of her bilateral upper and lower extremities, and diffuse abdominal tenderness.
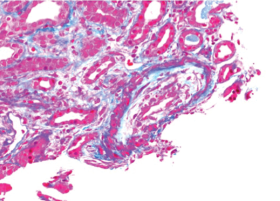
Figure 2. There is necrosis within the media layer of the vessel which is highly indicative of scleroderma renal crisis.
Laboratory tests revealed severe anemia, leukocytosis, severe thrombocytopenia, transaminitis and acute kidney injury with both hematuria and proteinuria (see Table 1). In addition, the LDH was elevated (1,394 IU/L, see Table 1), and the peripheral smear demonstrated multiple schistocytes. Nephrology and hematology specialists were consulted. Due to the patient’s acute presentation, there was a great concern for thrombotic thrombocytopenic purpura (TTP).
One day after admission, plasmapheresis was initiated. Despite this treatment, the patient’s renal function continued to deteriorate, and her creatinine increased from 2.5 to 3.7 mg/dL. When her ADAMTS13 level was noted to be elevated (77%), our suspicion for atypical TTP was raised.
The patient continued to be confused, weak and icteric. She also complained of pain in multiple joints, especially her hands. At this time, the rheumatology service at our hospital was consulted.
On the rheumatology clinical examination, this patient had sclerodactyly, extensive thickening of the skin on her face, the skin from her hands to above her elbows and the skin from her feet to her thighs. She had contractures of the hands, and she couldn’t make a full fist. Telangiectasia were noted over her face. There was no periungual erythema, capillary dilatations or calcinosis.