Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disease of immune dysregulation characterized by unchecked inflammatory responses leading to end-organ dysfunction. Primary HLH results from inherited mutations that impair the capacity for immune regulation; secondary HLH arises from the inappropriate response to an immune stimulus, such as infection, malignancy or autoimmunity.
What is less well known is whether patients with inborn metabolism errors can present with secondary HLH as an initial manifestation of their underlying metabolic disease.
The Case
A 9-month-old boy with a history of gross motor delay presented following three days of fever, vomiting and diarrhea. His initial evaluation was notable for transaminitis, coagulopathy and elevated serum ammonia, prompting transfer to the medical intensive care unit (MICU) for evaluation of acute liver failure.
He was febrile, tachycardic and ill-appearing on exam, with macrocephaly and hepatomegaly. He did not have splenomegaly, rash or arthritis.
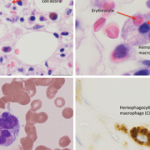
His repeat lab evaluation was notable for a markedly elevated ferritin of 29,930 ng/mL, elevated triglycerides at 210 mg/dL, anemia and thrombocytopenia, along with persistent transaminitis, coagulopathy and hyperammonemia. Liver and bone marrow biopsies demonstrated erythrophagocytosis consistent with HLH.
The patient’s immunologic evaluation was notable for hypogammaglobulinemia and neutropenia. Serum CD25 levels and natural killer (NK) cell functional studies (CD107a mobilization and perforin/granzyme B expression) were normal. He also had an extensive infectious evaluation of his blood, urine, stool, spinal fluid and respiratory secretions, none of which identified a pathogenic organism.
The patient was initially treated with ammonia-scavenger therapy and fresh frozen plasma (FFP) for coagulopathy prior to his liver and bone marrow biopsies. Within 24 hours after starting FFP, his ferritin level declined by over 50% and his coagulation studies improved.
Although our patient met five out of the eight diagnostic criteria for HLH, because of the dramatic improvement in his serum ferritin after just one dose of FFP, the decision was made to closely monitor him prior to starting stronger immunosuppressive medications.
Patients with inborn metabolism errors [may] present with secondary HLH as an initial manifestation of their underlying metabolic disease.
He did receive a 1 g/kg dose of intravenous immunoglobulin (IVIG) after he recrudesced with fever, but with continued improvement in his serum ferritin. He developed generalized seizures toward the end of his IVIG infusion. It was unclear whether the seizures were a result of HLH, the IVIG itself or a possible inborn metabolism error.
Therefore, he underwent magnetic resonance imaging (MRI) of his brain, which was normal. He was treated with 10 mg/kg of dexamethasone per day, which was tapered as his hyperammonemia, transaminitis and fever improved.
Our patient underwent concurrent biochemical and genetic evaluation for both primary HLH and inborn metabolism errors. An analysis of serum transferrin isoforms was suggestive of a type II congenital disorder of glycosylation (CDG). Rapid whole exome sequencing confirmed his presumed diagnosis by identifying compound heterozygous mutations in the COG4 gene that encodes for part of an oligomeric protein complex involved in Golgi apparatus structure and function.