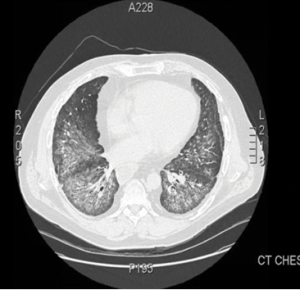
Figure 1.
This computed tomography of the chest shows bilateral, diffuse, ground-glass opacities and nodular opacities with intralobular septal thickening.
Antisynthetase syndrome (AS) is strongly associated with the presence of antibodies to aminoacyl-transfer RNA (tRNA) synthetases (ARSs) that are implicated in the pathogenesis of myositis and interstitial lung disease (ILD).
Antibodies against eight antisynthetases have been identified and are detected in 16–26% of patients with idiopathic inflammatory myopathies (IIM).1 Serum assays for five of these antibodies are commercially available: anti-histidyl (Jo-1), anti-threonyl (PL-7), anti-alanyl (PL-12), anti-isoleucyl (OJ) and anti-glycl (EJ). Anti-Jo-1 antibodies are seen more commonly in patients with polymyositis and dermatomyositis compared with the others that are found less frequently.2 These antibodies usually target the enzymatic component of tRNA synthetases; only anti-PL-12 recognizes tRNA directly.

