Phosphofructokinase deficiency (GSD type VII; PFK deficiency or Tarui disease) might be underdiagnosed because it presents with nonspecific exercise intolerance and myalgias. Until recently, Tarui disease could only be diagnosed by functional enzyme testing requiring muscle biopsy. The diagnostic evaluation has been simplified with the recent availability of gene testing. The remaining disorders are quite rare, with the exception of McArdle’s disease (GSD type V), and typically produce dynamic symptoms. Given the relatively common occurrence and characteristic clinical presentation, we will discuss McArdle’s disease in more detail here.
Case 1
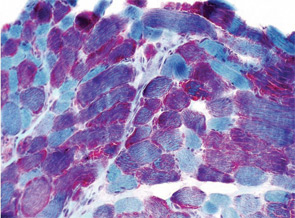
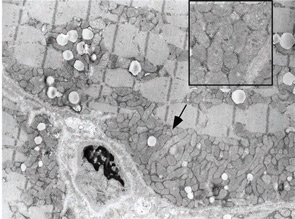
A 22-year-old male presented to the neuromuscular clinic with exertional myalgia and recurrent myoglobinuria. When the patient was a young child, his first symptoms were painful, transient contractures of his forearm finger flexors noted when climbing trees. Attacks became more frequent with age, and he first noticed dark urine associated with muscle cramping as a high school student. He did not report symptomatic improvement following short periods of rest. Physical examination was normal in clinic. There was no weakness, and no fasciculations or fixed contractures. His medical history and family history were noncontributory. Laboratory examination revealed an elevated CK (8,404 U/L; normal, 30–220 U/L), mild elevations of aspartate aminotransferase (75 U/L; normal, 15–41 U/L), and alanine aminotransferase (82 U/L; normal, 17–63 U/L). Urinalysis was negative for myoglobin. Routine muscle histology was unremarkable, but histochemical stains were not performed. EMG demonstrated mild myopathic changes. Molecular genetic testing revealed homozygosity for the common p.R49X mutation in the myophosphorylase (PYGM) gene.


