Evidence Regarding Therapy Is Lacking
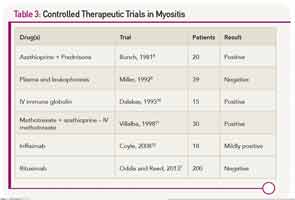
There are scant evidenced-based data to refer to when making therapeutic decisions for myositis (see Table 3). This may be one of the reasons why the prognosis for patients with polymyositis and dermatomyositis has not changed since the introduction of corticosteroids in 1949. About 20% to 40% of treated patients can enter a period of sustained remission, with the remaining 60% to 80% experiencing a continuous or fluctuating course.6 A concomitant malignancy, or the presence of circulating anti-SRP antibodies, suggest a poor prognosis. In most other situations, one cannot readily predict which patients will be good or poor responders.
Because idiopathic inflammatory myopathies are rare diseases, often have a very insidious onset (especially for inclusion body myositis), and can be challenging to diagnose, there have been few sizeable clinical trials. All but one of the published controlled trials had fewer than 50 patients. That study, the Rituximab in Myositis (RIM) trial, enrolled more than 200 patients from more than 30 centers. Despite the fact that more than 80% of subjects met the definition of improvement, the primary endpoints were not met.7
In the future, therapy may be guided by genetic or molecular testing. For example, rituximab may be more effective in patients with selected genetic signatures. Similarly, anti–interferon-α antibody (a biologic agent under development) may be effective in patients whose muscle has evidence for increased levels of interferon-α inducible genes. In the meantime, therapy for polymyositis and dermatomyositis remains empiric.

