Over the past two decades, exciting research has provided an important new perspective on the biology of bone in health and disease, revealing an active and sometimes loud crosstalk between bone and the immune system. This perspective is a radical departure from the traditional view of the relationship of these tissues. In the traditional view, the bone and immune system represented separate functional realms despite their close physical relationship in the bone marrow. There, the bone houses immune cells and provides a sanctuary for key events in immune cell development and differentiation. Many of the important discoveries on the interactions between bone and the immune system arose from studies on arthritis, where immune cells and bone are closely juxtaposed in the joint microenvironment.1–4 The dimension and variety of these interactions, however, were unexpected and have spurred the creation of a dynamic new field of research.
From a clinical perspective examining pathogenesis, bone seems to be the target and victim from damage inflicted at the hands of the immune system. Indeed, in inflammatory arthritis, bone erosion is the dreaded outcome of the immune system’s destructiveness. As is now realized, however, this view is too simplistic because bone can drive the immune system as well as withstand its blows. Evidence for an active role of bone in the pathogenesis of autoimmune diseases, such as systemic lupus erythematosus and inflammatory arthritis, has accumulated rapidly. This evidence includes provocative new findings on the properties of bone marrow niches, which are specialized microenvironments for the maturation of immune cells and for the maintenance of immunological memory. This new research field is called osteoimmunology and addresses the mutual interactions of bone and the immune system.5
Although pioneering works in the late 1970s identified potential interactions between bone and the immune system, the field truly emerged from landmark studies in the late 1990s. These studies demonstrated that T lymphocytes can trigger bone loss by inducing differentiation of osteoclasts.4,6 In this article, we will discuss current concepts of osteoimmunology in the context of rheumatologic disease. We will focus on the processes of bone degradation and formation that are regulated by the immune system in inflammatory arthritides. We will also consider the role of bone and bone marrow as a niche for immune cells, a mostly unknown yet important facet for the interplay of bone and immune systems in the pathogenesis of autoimmune disease.
RANKL has been established as a key mediator of inflammatory bone loss and one of the key molecules in the interactions that characterize osteoimmunology.
Osteoclasts, Osteoblasts, and Inflammatory Cells
Bone is living tissue and not just an inert or rigid framework to organize other tissues in the body. Bone grows, heals, transforms, and constantly remodels as it adapts to external mechanical forces. Adult vertebrates replace approximately 10% of their total bone content per year, with adolescents showing even higher turnover rates. The cells mediating these dynamic processes are the osteoclasts that degrade bone and the osteoblasts that produce new bone. A tightly regulated balance of osteoclastic and osteoblastic activity maintains a steady state of total bone mass in healthy adults. Diseases affecting bone, however, can disturb this balance and lead to both the loss and gain of bone tissue.7
Immune-osteoclast Interactions
To remodel bone efficiently, osteoclasts and osteoblasts interact extensively; in fact, osteoblasts are important regulators of osteoclast differentiation. Elegant in vitro systems have demonstrated that bone marrow–derived monocytes/macrophages differentiate into osteoclasts in the presence of osteoblast precursors or other mesenchymal cells such as synovial fibroblasts.4 To induce osteoclastogenesis, osteoblast precursors and synovial fibroblasts release soluble factors, including macrophage colony–stimulating factor, and express surface molecules, such as the long-sought osteoclast differentiation factor (see Figure 1, p. 37).1,4 When cloned in 1998, the osteoclast differentiation factor surprisingly showed identity with a protein called the receptor activator of nuclear factor–κB ligand (RANKL).8,9 RANKL had already been known for its role in the immune system, particularly for lymph node development, demonstrating clearly shared signalling mechanisms between bone and the immune system.10
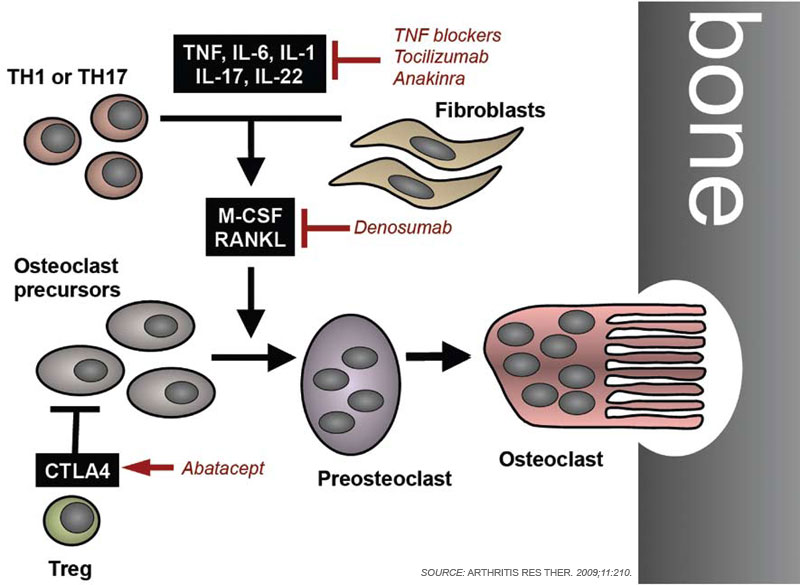
Since its discovery, RANKL has been established as a key mediator of inflammatory bone loss and one of the key molecules in the interactions that characterize osteoimmunology. When expressed on osteoblast precursors, synovial fibroblasts, and immune cells (see below), RANKL can bind to its receptor RANK on osteoclast precursors and induce osteoclastogenesis via intracellular signaling by NF-κB and the activation protein (AP)–1 transcription factor family.1,4 Studies from mouse models have provided important insights into the role of RANKL in bone. For example, mice deficient for RANKL show an osteopetrotic phenotype and a defective development of osteoclasts and low bone resorption. Interestingly, the expression of RANKL in the body is induced by proinflammatory cytokines, in particular tumor necrosis factor (TNF) as well as interleukin (IL)-1 and IL-17; this pattern of regulation further highlights the involvement of the immune system and inflammation in RANKL-mediated effects on bone.1,11