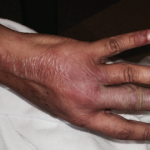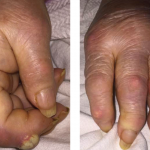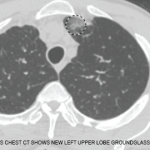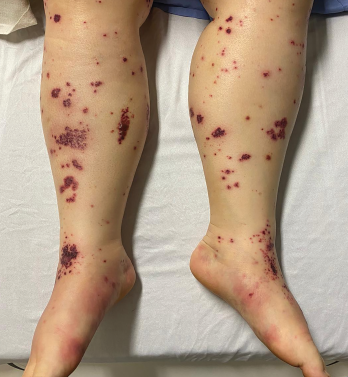
Figure 2. The patient had palpable purpura and hemorrhagic vesicles of both legs, with tender erythematous plaques and swelling over the feet.
Dermatologic findings can present in 40–60% of patients with EGPA, with palpable purpura being the most common manifestation.1 Non-hemorrhagic bullae are an exceedingly rare cutaneous manifestation of EGPA. Four cases we found described bullae as the initial cutaneous manifestation of EGPA, one as a relapsing feature and another in a patient with overlapping EGPA and cryoglobulinemic vasculitis.2-5,7,8 EGPA is typically not considered in the differential diagnosis of patients presenting with non-hemorrhagic bullae unless other clinical features are present.
Interestingly, four cases of EGPA-Wells syndrome overlap presented with non-hemorrhagic bullae.9 Wells syndrome is a rare form of non-infectious cellulitis with histological findings of eosinophilic dermal infiltration and flame figures (i.e., collagen fibers coated with eosinophilic degranulation products). All of the cases had clinical features of both Wells syndrome and EGPA. Our patient did not present with pseudocellulitis, nor did she have flame figures on biopsy.
In Sum
We describe a rare case of EGPA presenting as a non-hemorrhagic bullous eruption with peripheral eosinophilia. The patient was initially thought to have either a drug reaction or a primary immunobullous disorder, and this drove initial investigations and management. EGPA was suspected only after she developed microvascular hemorrhages and purpura. Although rare, EGPA should be kept in the differential diagnosis of patients presenting with non-hemorrhagic bullae if prodromal features of EGPA, such as adult-onset asthma and otolaryngologic symptoms, are present.
 Jordan Friedmann, MD, is a PGY-3 internal medicine resident at the University of British Columbia, Vancouver, and is planning on pursuing a career in rheumatology.
Jordan Friedmann, MD, is a PGY-3 internal medicine resident at the University of British Columbia, Vancouver, and is planning on pursuing a career in rheumatology.
 Julia Tan, MD, is a PGY-5 rheumatology resident at the University of British Columbia.
Julia Tan, MD, is a PGY-5 rheumatology resident at the University of British Columbia.
 Danny Mansour, MD, is a PGY-4 dermatology resident at the University of British Columbia. He is the co-chair of the Canadian Dermatology Association Resident and Fellows Society, where he has organized and led nationwide dermatology curricula. He is the chief resident for the January–June 2022 term and has a strong interest in medical education.
Danny Mansour, MD, is a PGY-4 dermatology resident at the University of British Columbia. He is the co-chair of the Canadian Dermatology Association Resident and Fellows Society, where he has organized and led nationwide dermatology curricula. He is the chief resident for the January–June 2022 term and has a strong interest in medical education.
 Sheila Au, MD, FRCPC, is a clinical associate professor in the Department of Dermatology and Skin Science at the University of British Columbia. She is an active member of the Canadian Dermatology Association’s Canadian Rheumatology and Dermatology Working Group and served as an associate editor of the Journal of Cutaneous Medicine and Surgery. Dr. Au received the 2019 Practitioner of the Year Award from the Dermatology Society of British Columbia.
Sheila Au, MD, FRCPC, is a clinical associate professor in the Department of Dermatology and Skin Science at the University of British Columbia. She is an active member of the Canadian Dermatology Association’s Canadian Rheumatology and Dermatology Working Group and served as an associate editor of the Journal of Cutaneous Medicine and Surgery. Dr. Au received the 2019 Practitioner of the Year Award from the Dermatology Society of British Columbia.
 Neda Amiri, MD, FRCPC, is clinical assistant professor in the Division of Rheumatology at the University of British Columbia, as well as the director of the PReDICT (Pregnancy and Rheumatic Disease Clinic) clinic at Mary Pack Arthritis Centre and a consultant in the Lupus Clinic. She attended internal medicine and rheumatology training at the University of British Columbia.
Neda Amiri, MD, FRCPC, is clinical assistant professor in the Division of Rheumatology at the University of British Columbia, as well as the director of the PReDICT (Pregnancy and Rheumatic Disease Clinic) clinic at Mary Pack Arthritis Centre and a consultant in the Lupus Clinic. She attended internal medicine and rheumatology training at the University of British Columbia.
Acknowledgments
We thank Hamid Masoudi, MD, FRCPC, staff pathologist at St. Paul’s Hospital, Vancouver, for his assistance reviewing the pathology.
References
- Micheletti RG, Chiesa Fuxench Z, Craven A, et al. Cutaneous manifestations of antineutrophil cytoplasmic antibody–associated vasculitis. Arthritis Rheumatol. 2020 Oct;72(10):1741–1747.
- Ishibashi M, Kudo S, Yamamoto K, et al. Churg-Strauss syndrome with coexistence of eosinophilic vasculitis, granulomatous phlebitis and granulomatous dermatitis in bullous pemphigoid-like blisters. J Cutan Pathol. 2011 Mar;38(3):290–294.
- Ng WC, Chen Y-C, Su Y-L. Zosteriform bullous eruption in a patient with antineutrophil cytoplasmic antibody‐negative eosinophilic granulomatosis with polyangiitis. Int J Dermatol. 2020 Jan;59(1):e6–e8.
- Mroz RM, Korniluk M, Swidzinska E, Chyczewska E. Churg-Strauss syndrome: A case report. Eur J Med Res. 2010 Nov 4;15 Suppl 2:92–94.
- Marra AM, Barilaro G, Villella V, Granata M. Eosinophilic granulomatosis with polyangiitis (EGPA) and PRES: A case-based review of literature in ANCA-associated vasculitides. Rheumatol Int. 2015 Sep;35(9):1591–1595.
- de Groot K, Harper L, Jayne DRW, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody–associated vasculitis: A randomized trial. Ann Intern Med. 2009 May 19;150(10):670–680.
- Abeyaratne DDK, Liyanapathirana C, Fonseka C, Wijekoon P. A rare case of eosinophilic granulomatosis with polyangiitis associated with cryoglobulinemia presenting with a bullous skin eruption of the lower limbs. Case Rep Med. 2018 Jan 16;2018:3124281.
- Sherman S, Gal N, Didkovsky E, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) relapsing as bullous eruption. Acta Derm Venereol. 2017 Mar 10;97(3):406–407.
- Fujimoto N, Wakabayashi M, Kato T, et al. Wells syndrome associated with Churg-Strauss syndrome. Clin Exp Dermatol. 2011 Jan;36(1):46–48.
- Borrego L, Maynard B, Peterson EA, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. 1996 Mar;148(3):897–909.
- Naito K, Morioka S, Ogawa H. The pathogenic mechanisms of blister formation in bullous pemphigoid. J Invest Dermatol. 1982 Nov;79(5):303–306.