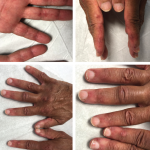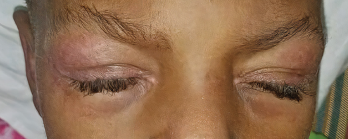
Photo 1: The patient presented with heliotrope rash.
Clinically amyopathic dermatomyositis (CADM), a rare subset of dermatomyositis (DM), is an autoimmune disease characterized by cutaneous findings of typical DM without evidence of myositis. Childhood presentation of CADM is rare, and not many studies describe the epidemiology of juvenile CADM.1,2
Although lung disease is rare among patients with juvenile DM, a few reports have been published since 2007 about interstitial lung disease (ILD) in children with CADM.3 Rapidly progressive ILD occurs more in CADM patients than in classic DM patients and has a poor prognosis.