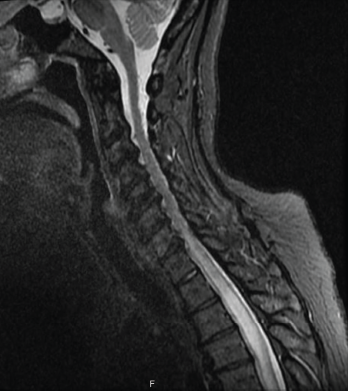
Figure 1: MRI of the spine
Sjögren’s syndrome is a chronic multi-system autoimmune disease characterized by inflammation and subsequent destruction of exocrine glands. Sjögren’s syndrome can present with glandular or extra-glandular manifestations. Neuromyelitis optica spectrum disorder (NMOSD) is a rare central nervous system (CNS) autoimmune disease that can present as the initial manifestation in less than 5% of patients with Sjögren’s.
It is important to recognize relationships between autoimmune diseases, especially Sjögren’s syndrome and NMOSD. This rare association requires a high degree of suspicion in a patient with Sjögren’s syndrome with a suggestive clinical presentation and imaging findings.