What’s Your Diagnosis?
- Majocchi’s granuloma/tinea infection
- Granuloma annulare
- Palisaded neutrophilic granulomatous dermatitis
- Familial sarcoidosis/Blau syndrome
Correct answer: D
Familial Sarcoidosis/Blau Syndrome
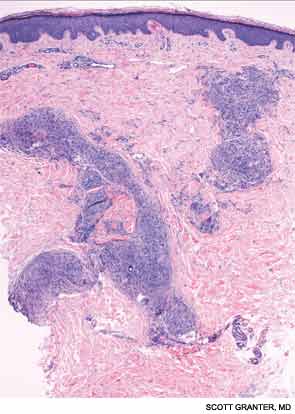
A skin biopsy of a representative lesion was performed, and histopathologic examination revealed nodular aggregates of histiocytes forming non-necrotizing, “naked” granulomas (see Figure 2). Special stains and PCR studies for infectious organisms including mycobacteria were negative. Genetic testing revealed that our patient and multiple family members had a base substitution in the CARD15/NOD2 gene, consistent with a diagnosis of Blau syndrome. Topical steroids were prescribed for her lesions, which resolved with treatment.
Blau syndrome was first described in 1985 by Edward Blau, a pediatrician. It’s a rare, autosomal dominant autoinflammatory disorder with a typical onset within the first two decades of life. Clinically, patients most commonly present with a triad of granulomatous dermatitis, symmetric arthritis and recurrent uveitis. Patients may also have other organ involvement, including hepatic, renal, cardiovascular or neurologic complications.1,2 Mutations that cause Blau syndrome have been mapped to CARD15/NOD2, a caspase recruitment domain gene.
The CARD15/NOD2 gene contains three domains. It is most commonly known for mutations in the third domain, associated with Crohn’s disease, which involves granulomatous inflammation of the bowel. Mutations in the second domain are associated with both Blau syndrome and early onset sarcoidosis. CARD15/NOD2 activates NF-kB in the setting of bacterial cell wall components.3 In Blau syndrome, genetic mutations in the second domain are usually gain-of-function mutations, leading to increased NF-kB activation and possibly increased activity of other inflammatory cytokines.4
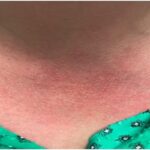
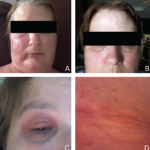
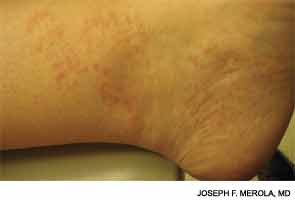
Blau syndrome often presents earliest and most commonly in the skin. Patients present with multiple erythematous and waxy dermal papules and plaques with a lack of scale. Skin biopsy is helpful in diagnosis because it often demonstrates granulomatous inflammation.5
Discussions of Other Choices
Majocchi’s granuloma occurs when a dermatophyte infection involves the deeper structures of the skin, such as the hair follicles and the dermis. These lesions occur commonly in areas with hair follicles, such as the legs. Patients can also demonstrate other evidence of dermatophyte infection, such as onychomycosis or tinea pedis. Histopathologic examination of Majocchi’s granuloma would reveal fungal forms on the biopsy.