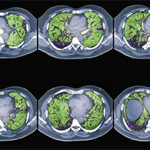
Classification of CTD-ILD Based on Histopathology
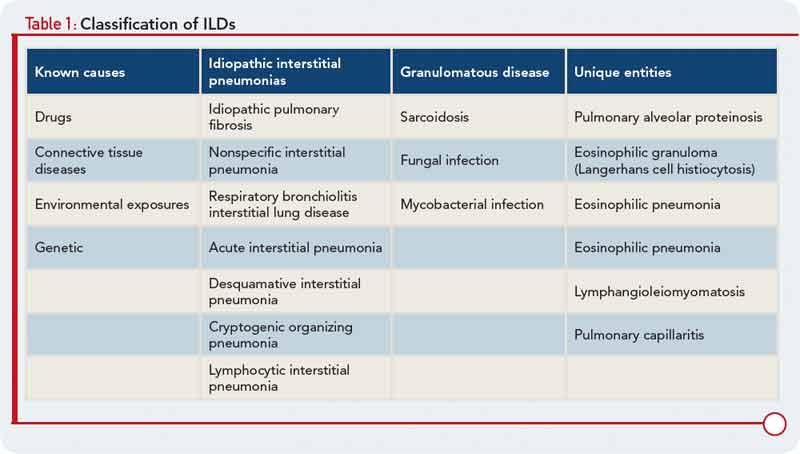
To characterize the histologic patterns in surgical lung biopsy (SLBx) specimens from patients with ILD, including those with CTD-ILD, pathologists use the classification scheme developed for the idiopathic interstitial pneumonias (IIPs).3 The IIPs are a group of seven ILDs of unknown cause, with potentially overlapping clinical and radiological features but different prognoses (see Table 1). Over the years, a challenging nomenclature and ever-changing classification scheme has created confusion around the IIPs. Also clouding understanding is the fact that, although the seven histologic patterns decisively distinguish one IIP from another, the patterns are not specific to “idiopathic” disease and may occur in ILD of any cause. For example, the fibrotic pattern of usual interstitial pneumonia (UIP pattern) has a number of potential causes, including chemotherapy-induced ILD, hypersensitivity pneumonitis, sarcoidosis, or any of the CTDs. The UIP pattern is the most common histological pattern in patients with IIP. However, among patients with CTD-ILD who undergo SLBx, the pattern of nonspecific interstitial pneumonia (NSIP pattern) is most frequently identified. It is unclear whether the NSIP pattern appears to predominate because of significant selection bias: relatively few patients with CTD-ILD undergo SLBx.
Among surgically biopsied patients with SSc-associated ILD (SSc-ILD), the NSIP pattern is most common, identified in 64–78% of SSc-ILD subjects in various studies.4-6 All of these studies are potentially affected by selection bias. Similarly, in small (also obviously biased) case series, the NSIP pattern predominated among surgically biopsied subjects with PM/DM, primary Sjögren’s, and mixed connective tissue disease. In contrast, although also based on limited data, studies from several investigators suggest that the UIP pattern predominates in RA-associated ILD (RA-ILD).7,8
Results from several studies have shown that, among those with IIP, it is the histologic pattern that drives prognosis. While controlling for other potentially important predictors, patients with UIP pattern have worse survival than patients with NSIP (or any other) pattern lung injury. What is far less clear, and a focus of much debate and ongoing research, is to what degree the histopathologic pattern identified in SLBx specimens from patients with CTD-ILD impacts prognosis. Bouros and colleagues studied 80 subjects with SSc-ILD who had undergone SLBx and found that five-year survival was not significantly different between those with UIP and those with NSIP-pattern histology. Rather, mortality was associated with lower initial diffusing capacity of the lung for carbon monoxide, lower initial forced vital capacity (FVC), and a decline in diffusing capacity over time.4 Similarly, Park and colleagues studied survival in 93 individuals with CTD-ILD who underwent SLBx (37 with SSc-ILD, 28 with RA-ILD, 11 with Sjögren’s-associated ILD, eight with myositis-associated ILD, and nine others).8 They found that age, duration of dyspnea, and FVC were associated with survival, but histological pattern (i.e., NSIP pattern vs. UIP pattern) was not. Results from other studies that included subjects with CTD-ILD suggest the presence of fibrosis (i.e., UIP pattern) is associated with worse survival.9
Because of the conflicting data, many clinicians are uncertain about whether and when to have their patients with CTD-ILD undergo SLBx. On the one hand, many physicians believe that, as is the case with the IIP, UIP-pattern histology confers a worse prognosis than NSIP-pattern histology among patients with CTD-ILD. Physicians holding this view would have their CTD-ILD patients undergo SLBx because the results could better define disease trajectory. On the other hand, some clinicians choose not to send their CTD-ILD patients for SLBx, either because they are not convinced that the histological pattern trumps pulmonary physiology in determining prognosis, or more likely because they don’t expect the results to change management: they plan to treat (or perhaps already are treating) their CTD-ILD patients with immunosuppressive medications, and the pattern identified in SLBx specimens would not affect their choice of therapeutic agent or anticipated duration of therapy. There is greater consensus about the utility of a surgical lung biopsy in patients with CTD-ILD when an etiology for the ILD other than CTD (e.g., hypersensitivity pneumonitis) is seriously considered or when imaging features suggest malignancy or infection (e.g. progressive nodules, cavitation, consolidation, pleural thickening, or effusion).
In addition to the primary histologic pattern within the interstitium, certain microscopic features suggest the presence of “autoimmune lung disease.” These include dense perivascular collagen, intense plasmacytic infiltration, extensive pleuritis, and numerous lymphoid aggregates with germinal center formation. Whether or exactly how these features affect response to immunomodulatory therapy or prognosis has yet to be definitively determined.
What Does High-Resolution Computed Tomography Tell Us?
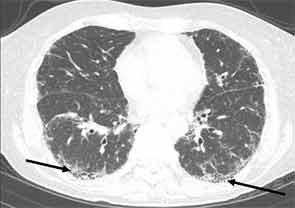
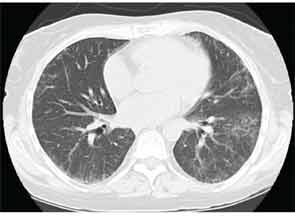
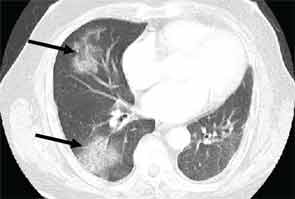
High-resolution computed tomography (HRCT) is the imaging modality of choice to assess ILD and can accurately predict the underlying histological pattern in many cases (see Figures 1–3). For example, when lower zone-, posterior-, subpleural-predominant reticular opacities with traction bronchiectasis and honeycombing are identified on HRCT in the absence of significant ground glass opacities, the certainty of finding a histologic UIP pattern by SLBx exceeds 96%.10 In fact, when those features are present, the HRCT is considered to display “a radiologic UIP pattern.” Similarly, radiologists often use the term “radiologic NSIP pattern” to describe a HRCT that shows lower zone–predominant ground glass opacities (either alone or in combination with reticular opacities) but no honeycombing. Because of the high degree of accuracy for predicting UIP-pattern histology, when a radiologic UIP pattern is present (in patients with either idiopathic disease or CTD-ILD) on HRCT, SLBx is not performed. However, pattern determination by HRCT is far less accurate for the other, non-UIP, patterns of ILD. In those cases, the decision about whether to biopsy or not, particularly among patients with CTD-ILD (as described above) is less straightforward.
For patients with RA-ILD, a radiologic UIP pattern is a poor prognostic marker. Kim and colleagues studied 82 RA-ILD subjects over a median five years of follow-up. They observed that subjects with a radiologic UIP pattern on HRCT had worse survival than those without radiologic UIP patterns (3.2 years vs 6.6 years, p=0.04).11 In light of these data, the authors have proposed that incorporating knowledge of underlying HRCT pattern in RA-ILD should inform the management approach: Those with RA-NSIP should be treated with immunosuppression and those with RA-UIP should be counseled as to their more unfavorable prognosis and considered for lung transplantation. Additional studies are needed to determine with greater confidence the role of HRCT in predicting prognosis in patients with CTD-ILD.
Defining “CTD-ILD”
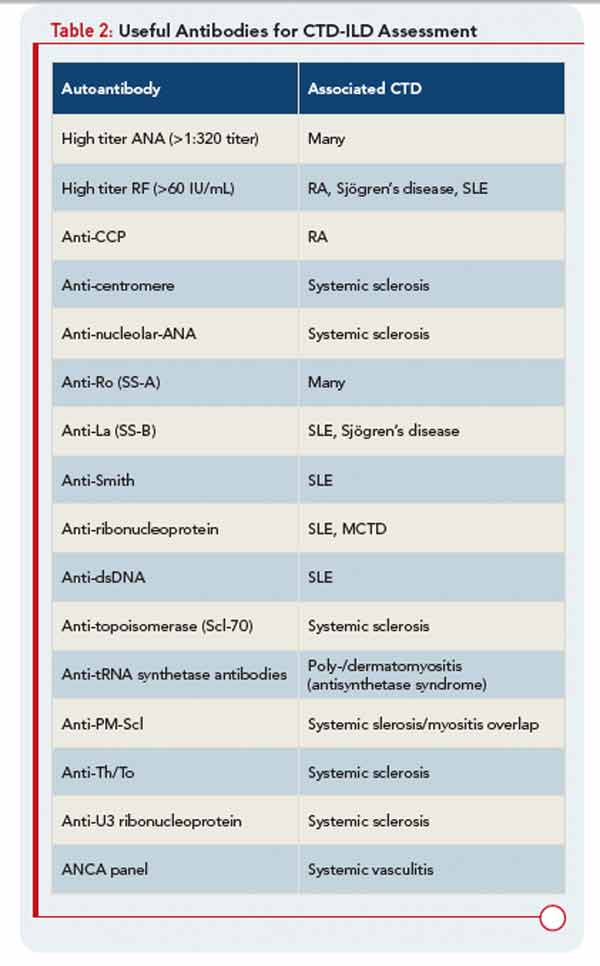
In the patient with CTD, determining whether ILD is, in fact, CTD-ILD or ILD of some other etiology is a process of elimination. Alternative etiologies including respiratory infection, medication toxicity, environmental exposure, and recurrent aspiration must be excluded prior to diagnosing CTD-ILD. Complicating matters is the fact that ILD may be the presenting manifestation of CTD, with joint, skin, or other organ involvement appearing months or years after ILD. Nearly 15% of individuals presenting with what is initially diagnosed as idiopathic ILD (i.e., IIP) will eventually develop a classifiable CTD; thus, clinicians should be on the lookout for features of CTD in all their patients with “idiopathic” ILD. Confirming that ILD is a manifestation of CTD may impact management and prognosis thus a thorough assessment for underlying CTD is warranted in patients with idiopathic interstitial pneumonia. Yet there is no standardized approach for such assessments. In our opinion, the evaluation should include a thorough history and physical examination, and measuring for specific autoantibodies (see Table 2). The process can be optimized by encouraging a multidisciplinary approach that includes rheumatologic consultation.
Implications of a CTD-ILD Diagnosis
There are numerous implications of a CTD-ILD diagnosis. Most significantly, some data suggest that for a given histological pattern, subjects with CTD-ILD have a more favorable prognosis than subjects with idiopathic ILD. Other potential clinical implications of diagnosing a CTD include the following: 1) a CTD diagnosis could provide a context for extrathoracic disease manifestations (e.g., esophageal dysmotility); 2) if a CTD is diagnosed, the need for and timing of surveillance for certain extrathoracic features (e.g., pulmonary hypertension) is better defined; and 3) diagnosing a CTD may guide therapeutic decision making. As for research aimed at advancing the field of CTD-ILD, precisely phenotyping patients allows for greater validity of—and the ability to compare results across—studies of the pathobiology and natural history of CTD-ILD.
It is easy to confidently diagnose CTD—more specifically, RA—as the cause for the ILD in a 60-year-old male, former smoker with bilateral wrist synovitis, a high-titer anticyclic citrullinated peptide antibody, and a radiologic UIP pattern on HRCT, but there are a large number of patients with some features of a CTD (but not enough to satisfy ACR classification criteria) and ILD in a radiological or histological pattern commonly found in CTD-ILD. For example, we frequently encounter patients with ILD who are suspected of having a CTD based on the presence of circulating autoantibodies, certain autoimmune histopathologic features in SLBx specimens, or finding subtle extrathoracic manifestations. However, current CTD classification schemes do not accommodate these individuals, so they are labeled as having idiopathic ILD by default. This leaves them in limbo: they have a “flavor” of CTD and thus (perhaps appropriately) do not qualify for trials enrolling subjects with IIP, but they don’t meet criteria for the diagnosis of a CTD either. For example, a patient with idiopathic ILD and UIP-pattern histology would not be allowed in most therapeutic trials for IPF if, in addition to the UIP pattern, the SLBx revealed numerous lymphoid follicles with germinal centers; this is because the lymphoid follicles raise suspicion that such a patient may have CTD-ILD (even though they fulfill no ACR criteria for CTD) rather than IPF.
Further, despite the knowledge that ILD occurs commonly as a manifestation of CTD, current classification schemes—other than for SSc—do not consider ILD as a diagnostic criterion for CTD. Some experts have speculated that most patients with IIP (especially those with radiological or histological NSIP patterns) have an underlying CTD. This is because NSIP is the most common pattern in patients with CTD. Added to that, many patients with IIP have circulating autoantibodies, even those with reportedly high specificity for CTD (e.g., anticyclic citrullinated peptide). Song and colleagues observed that in subjects with IIP, the presence of circulating autoantibodies correlated with the presence of histologic features (mentioned above) of “autoimmune lung disease.”12
Current strategies for identifying and classifying patients with only some features of CTD are both controversial and inadequate. Proposed terms to describe them include “undifferentiated CTD,” “lung-dominant CTD,” and “autoimmune featured ILD.” Corte and colleagues retrospectively studied subjects with IIP who had undergone SLBx, 45 with NSIP- and 56 with UIP-pattern histology.13 They observed that features of CTD are common in patients with IIP: 31% of subjects with NSIP- and 13% of subjects with UIP-pattern histology fulfilled established diagnostic criteria for undifferentiated CTD (UCTD).14 Importantly, when a broader and less specific set of proposed criteria for UCTD was applied, they found an astounding 71% of subjects with NSIP-pattern histology and 36% of those with UIP could be “defined” as having UCTD!15 Because of its lack of specificity, they argued against further implementation of the broader set of UCTD criteria in patients with ILD. Moreover, although a diagnosis of UCTD was significantly associated with a pattern of NSIP, conveying a label of UCTD did not impact survival. Vij and colleagues considered subjects to have “autoimmune featured ILD” if they had a sign or symptom suggestive of a CTD—including at least one serologic marker—but did not meet criteria for definite CTD.16 Subjects with autoimmune featured ILD had survival rates no different from subjects with IPF, but worse than subjects with CTD-ILD. Interestingly, among subjects with autoimmune featured ILD, the presence of an antinuclear antibody with a titer of >1:1280 was associated with improved survival.
Well-organized prospective studies are needed to determine the true implications of finding serologic or histopathologic features of CTD in patients without a classifiable CTD. Productivity of such studies will be greatly enhanced if consensus is reached on terminology and a classification scheme for these patients.
Natural Progression of CTD-ILD
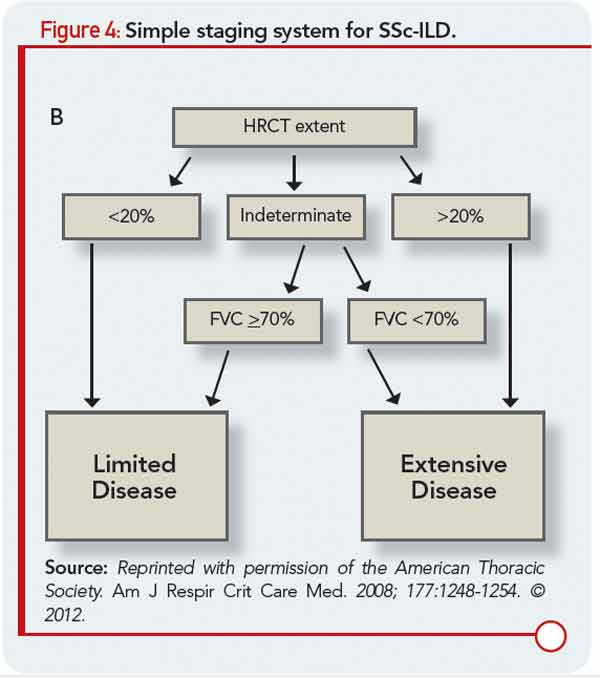
Like patients with other forms of ILD, patients with CTD-ILD most often present with the insidious onset and gradual progression of dyspnea. The heterogeneity among subjects has made large-scale natural history studies of ILD in RA, PM/DM, and each of the other CTDs challenging, so more intense research will be required to advance understanding of ILD behavior over time in those patients. In patients with SSc-ILD, a simple staging system (see Figure 4) has been recently proposed that incorporates HRCT and FVC, and results suggest it provides a more accurate prognostic separation across SSc-ILD than has been achieved with any single modality in isolation.17 We believe the application of this staging system could impact clinical management decisions as well as future clinical treatment trial design, although additional investigation of this staging system, and validation in additional study samples would add confidence in its wide-scale implementation. In contrast to initial beliefs, bronchoalveolar lavage (BAL) does not predict the likelihood of disease progression or treatment responsiveness in CTD-ILD, so its routine use as a diagnostic tool in patients evaluated for CTD-ILD should be discouraged. However, BAL is often indicated to exclude infection and may also be performed as a component of research protocols.
Management Dilemmas
Although there are few prospective data to guide therapeutic decision making, it is clear that not all patients with CTD-ILD require treatment. We believe whether to treat a patient with CTD-ILD depends on the severity and pace of the ILD and which comorbid conditions or contraindications to treatment exist. Therapy for CTD-ILD is generally reserved for patients with clinically significant, progressive disease. When considering immunomodulatory therapy options for CTD-ILD, both intra- and extrathoracic disease manifestations need to be taken into consideration. Because multiple systems may be affected by CTD, coordinated care among rheumatologists and other subspecialists can be helpful. Although extrathoracic manifestations may determine the initial immunomodulatory regimen, quite often, it is the ILD that dictates the intensity (i.e., specific agent and duration) of the immunosuppressive therapy.
We believe patients with CTD-ILD require a multidisciplinary approach to management, and the challenges in diagnosing, classifying, and treating CTD-ILD create opportunities ripe for carefully planned research.
We are in desperate need of better therapies for CTD-ILD. Determining which drugs are effective for patients with CTD-ILD will require the completion of thoughtfully constructed and carefully conducted large-scale therapeutic trials. Until then, we have to rely on data from small case series or retrospective studies, scientific rationale, and inferences drawn from the few controlled clinical trials conducted for SSc-ILD.
ILD is a serious, potentially life-threatening manifestation of CTD. Patients with all forms of CTD are at risk for developing ILD, and ILD may be the first or only manifestation of a CTD. There are numerous challenges and opportunities related to the clinical care and research of CTD-ILD. We believe patients with CTD-ILD require a multidisciplinary approach to management, and the challenges in diagnosing, classifying, and treating CTD-ILD create opportunities ripe for carefully planned research.
Disclosures
Drs. Fischer and Swigris are investigators for the NIH-funded Scleroderma Lung Study II. Dr. Fischer is a speaker, consultant, and advisory board member for Actelion pharmaceuticals and a speaker and advisory board member for Gilead pharmaceuticals.
Dr. Fischer is associate professor of medicine, acting chief of the division of rheumatology, and in the autoimmune and interstitial lung disease program at National Jewish Health in Denver. Dr. Swigris is associate professor of medicine and in the autoimmune and interstitial lung disease program at National Jewish Health.
References
- Fischer A, West SG, Swigris JJ, et al. Connective tissue disease-associated interstitial lung disease: A call for clarification. Chest. 2010;138:251-256.
- Frankel SK, Brown KK. Collagen vascular diseases of the lung. Clin Pulm Med. 2006;13:25-36.
- Joint Statement of the American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277-304.
- Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:1581-1586.
- Fischer A, Swigris JJ, Groshong SD, et al. Clinically significant interstitial lung disease in limited scleroderma: Histopathology, clinical features, and survival. Chest. 2008;134:601-605.
- Kim DS, Yoo B, Lee JS, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:121-127.
- Lee HK, Kim DS, Yoo B, et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest. 2005;127:2019-2027.
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: Idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705-711.
- Kocheril SV, Appleton BE, Somers EC, et al. Comparison of disease progression and mortality of connective tissue disease-related interstitial lung disease and idiopathic interstitial pneumonia. Arthritis Rheum. 2005;53:549-557.
- Hunninghake GW, Zimmerman MB, Schwartz DA, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164:193-196.
- Kim EJ, Collard HR, King TE, Jr. Rheumatoid arthritis-associated interstitial lung disease: The relevance of histopathologic and radiographic pattern. Chest. 2009; 136:1397-1405.
- Song JW, Do KH, Kim MY, et al. Pathologic and radiologic differences between idiopathic and collagen vascular disease-related usual interstitial pneumonia. Chest. 2009;136:23-30.
- Corte TJ, Copley SJ, Desai SR, et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J. 2012;39:661-668.
- Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): A review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17:615-620.
- Kinder BW, Collard HR, Koth L, et al. Idiopathic nonspecific interstitial pneumonia: Lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176:691-697.
- Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: A distinct entity. Chest. 2011;140:1292-1299.
- Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: A simple staging system. Am J Respir Crit Care Med. 2008;177:1248-1254.