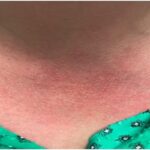
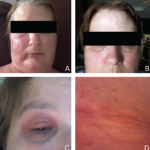
Differential Diagnoses
Infection: For the first few episodes of high fever and gastrointestinal (GI) symptoms, infection was a main concern. The patient had no recent travel history or animal exposure. Routine and specific tests for bacterial and viral etiology, as well as imaging studies, did not reveal any infection.
Allergic disorders: The patient’s medical history included intermittent asthma/bronchitis and rhinitis, and use of inhalers/nebulizers. She continued to have febrile episodes, with facial erythema, GI symptoms and myalgias. At this point, asthma and rhinitis focused us on allergic diseases, both common and uncommon, such as food-dependent, exercise-induced anaphylaxis and systemic mastocytosis (SM). However, skin testing and RAST assays, serum IgE, plasma IL-5 and tryptase levels all were negative or normal.
SM patients present with urticaria pigmentosa, lymphadenopathy, hepatosplenomegaly and/or patchy or diffusely increased tracer uptake on bone scanning.1 Serum tryptase level is elevated, and KIT mutation is detected in most patients with SM.2 Generally, fever and arthralgias are not seen in SM. The absence of the above manifestations argues against a diagnosis of SM in our case.
Carcinoid syndrome: This neuroendocrine tumor comprises symptoms of cutaneous flushing, diarrhea, cardiac valvular lesions, and bronchospasm related to excessive serotonin production.3 Symptoms of the syndrome are intermittent to progressive, and these symptoms usually do not occur until the primary tumor metastasizes. Clinical workup includes laboratory testing of 24-hour urinary excretion of the serotonin metabolites 5-hydroxyindoleacetic acid (5-HIAA) and plasma chromogranin A, and an imaging study. While our patient had some symptoms associated with carcinoid syndrome, her disease flares were intermittent and became less frequent and milder over time.4 In addition, fever is usually absent in carcinoid syndrome. Further, a CT scan of the chest, abdomen and pelvis, as well as endoscopies, did not disclose any signs of malignancy in the internal organs. In addition, her illness duration, relapsing and remitting course, and extensive workup made malignancy a less likely etiology.
Systemic autoimmune disease: After failure to substantiate a diagnosis of an allergic disorder, one would be concerned about a systemic autoimmune disease, such as systemic lupus erythematosus or vasculitis. However, there were no signs of any internal organ damage or cutaneous signs of vasculopathy (e.g., purpura, nodules, urticaria or ulcers). Moreover, complete serologic testing was negative. These data discount a diagnosis of a systemic autoimmune disease.
Inflammatory bowel disease (IBD) & celiac disease: Over the first two years, the patient continued to have episodic diffuse abdominal pain and explosive, non-bloody diarrhea, as well as diffuse myalgias, arthralgias affecting the knees and ankles and rash. Evaluation for IBD and celiac disease was completed. Esophagogastroduodenoscopy demonstrated gastritis, and a colonoscopy with tissue biopsies was unremarkable. CT of the abdomen/pelvis during acute attacks showed mesenteric signs of panniculitis (see Figure 2, right). A remote surgical history of appendectomy was noted, but there was no evidence of small bowel obstruction.
In IBD patients, lower extremity arthralgias/arthritis occurs in approximately 20%. Bloody diarrhea is common, and cutaneous findings more specific to IBD can be seen in 10% of patients, including erythema nodosum and pyoderma gangrenosum, whereas erythematous patches or plaques are astonishingly rare.5,6 Nevertheless, there was no endoscopic or tissue evidence of IBD in this patient. Dermatitis herpetiformis is more common in celiac disease, but was not seen in our patient.7 In addition to negative serology and duodenal pathology, genetic testing for celiac disease was negative, making a diagnosis of celiac disease unlikely.
Weber-Christian disease: Given the CT finding of abdominal panniculitis and fever, one would be concerned about Weber-Christian disease, though the disease concept is controversial. The disease is also known as relapsing febrile nodular nonsuppurative panniculitis. Subcutaneous nodules are common and prominent, but conspicuously absent from our patient.8 Generally, a CT finding of mesenteric panniculitis is a relatively common and nonspecific finding during evaluation of abdominal pain, and can be attributable to numerous causes.9 In our case, a repeat abdominal CT a month after resolution of her abdominal complaints showed disappearance of the radiographic finding.
Hereditary periodic fever syndromes: The patient continued to have episodic fever/chills, abdominal pain/diarrhea, and shortness of breath (asthmatic bronchitis), as well as rash and left knee pain/swelling. Within the first two years, three severe episodes prompted three separate hospitalizations. There was a small pericardial effusion on chest CT, but an echocardiogram was unremarkable. Her CRP was slightly elevated, and the ESR and SAA were normal. A detailed family history failed to identify evidence of a periodic fever syndrome. Regardless, a presumptive clinical diagnosis of familial Mediterranean fever (FMF) was made, and empiric oral colchicine 0.6 mg twice daily began. The patient initially felt better, but colchicine was eventually discontinued due to the lack of sustained efficacy. Subsequently, a targeted genetic analysis of MEFV did not demonstrate evidence of mutations associated with FMF.
Given the periodic nature of the disease course and lack of detectable autoantibodies in this case, we entertained a systemic autoinflammatory disease or periodic fever syndrome. Among these diseases are classic hereditary periodic fever syndromes.10
Typically, the FMF phenotype includes febrile episodes lasting less than 72 hours, lower extremity erysipeloid rash, and acute abdomen with more constipation than diarrhea.11 However, our patient did not present with the classic clinical characteristics, and complete sequencing of the MEFV gene failed to identify known mutations. Although she had a transient therapeutic response to colchicine, these data together argue against a diagnosis of FMF.
In cryopyrin-associated periodic syndrome (CAPS), also called NLRP3-associated autoinflammatory diseases, patients are often affected by generalized cold-induced urticaria, conjunctivitis and sensorineural hearing loss associated with NLRP3 mutations that may be revealed by gene sequencing.12-14
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) patients typically have abdominal pain, fevers lasting ~14 days, localized myalgias involving a group of muscles overlying eruptions, pleurisy, and periorbital edema, and TRAPS is associated with mutations of the TNFRSF1A gene.15
In hyper-IgD syndrome (HIDS) or mevalonate kinase (MVK) deficiency, disease onset occurs almost exclusively in children, and this disease shares similar clinical phenotype with other hereditary periodic fever syndromes. A more specific phenotype may consist of generalized lymphadenopathy, symmetric polyarthritis, and erythematous macules/papules. Genetic testing usually demonstrates MVK gene mutations.16
NOD2-associated diseases: The patient underwent a six-gene panel next-generation sequencing of her blood cells for periodic fever syndrome, and the molecular analysis was positive and heterozygous only for the NOD2 variants, IVS8+158 and R702W. Further laboratory study showed that blood CD4/CD8 T cell ratio was elevated, and B lymphocyte count was normal. Plasma cytokine levels were normal. Circulating vascular endothelial growth factor (VEGF) level was 604 pg/mL (normal <393 pg/mL), and intercellular adhesion molecule 1 (ICAM-1)/vascular cell adhesion molecule 1 (VCAM-1) was normal.
With identification of the NOD2 variants, we then focused our diagnosis on another subset of autoinflammatory diseases, which are associated with NOD2 variants.17 These diseases largely encompass Crohn’s disease, Blau syndrome and Yao syndrome.
Blau syndrome (BS) typically manifests as a triad of granulomatous dermatitis, uveitis and inflammatory arthritis. The disease primarily occurs in children as an autosomal dominant disorder, and adult-onset disease is extremely meager.18 Dermatitis commonly comprises erythematous papular scaly rash. Arthritis affects the wrist, knee, ankle and proximal interphalangeal joints (PIPs), leading to campytodactyly (flexion of PIPs) and rheumatoid arthritis-like and cystic articular changes.19 Both dermatitis and arthritis are nearly symmetric and persistent. Of note, fever and GI symptoms are unusual in the disease.19