Yao Syndrome (YAOS, OMIM617321)
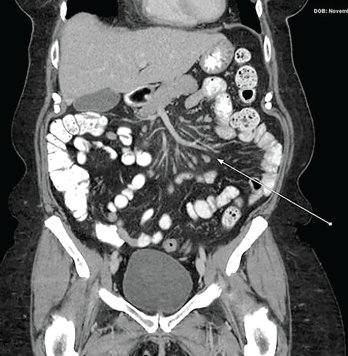
Figure 2: CT of the Abdomen & Pelvis with Contrast
The arrow shows findings of mesenteric panniculitis manifested as increase in density of the mesenteric fat surrounding the mesenteric vessels and increase in size and number of mesenteric lymph nodes.
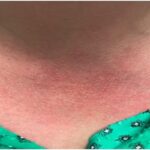
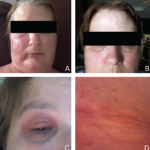
YAOS, formerly designated as NOD2-associated autoinflammatory disease, is characterized by periodic fever, dermatitis, arthritis and swelling of the distal extremities, as well as GI and sicca-like symptoms.6,20-22 Patients generally have flu-like symptoms, followed by episodic fever and erythematous patches or plaques with pathological finding of spongiotic dermatitis mostly. An episodic flare may last a few days to weeks with an asymptomatic interval of several weeks or months.
GI symptoms are common, including recurrent abdominal pain, cramping, bloating and non-bloody diarrhea, and the symptoms may last several days to weeks. There is no endoscopic, radiological or histopathological evidence of IBD. Oligo- or polyarthritis is common, and occurs periodically without joint erosions, destruction or deformities; joints appear normal when the disease is quiescent. Approximately 25% patients develop intermittent, unilateral or bilateral, distal lower extremity swelling involving the ankles and feet.23 In addition, patients can have oral ulcers and chest pain from pericarditis or/and pleuritis.