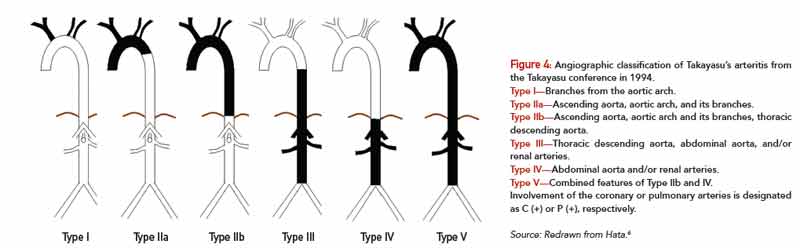
Type I—Branches from the aortic arch.
Type IIa—Ascending aorta, aortic arch, and its branches.
Type IIb—Ascending aorta, aortic arch and its branches, thoracic descending aorta.
Type III—Thoracic descending aorta, abdominal aorta, and/or renal arteries.
Type IV—Abdominal aorta and/or renal arteries.
Type V—Combined features of Type IIb and IV.
Involvement of the coronary or pulmonary arteries is designated as C (+) or P (+), respectively.
Source: Redrawn from Hata.6
Conventional angiography remains the gold standard for the diagnosis of TAK. MRA and CTA can also be used to evaluate large- and medium-size vessels. Rheumatic diseases that affect the aorta or its branches include giant cell arteritis, primarily differentiated from Takayasu’s by age of disease onset and distribution of lesions. Granulomatous vasculitis can be demonstrated on histologic examination in both diseases. Other rheumatic diseases, such as relapsing polychondritis, ankylosing spondylitis, Behçet’s disease and Cogan’s syndrome, may have a similar arteriographic appearance to TAK but can be differentiated on the basis of their other clinical manifestations. Infectious or “mycotic” aneurysms, atherosclerotic disease, fibromuscular dysplasia, congenital co-arctation and inherited disorders of connective tissue, such as Marfan’s syndrome and Ehlers Danlos, must also be considered.
The mainstay of therapy is suppression of inflammation and preservation of vascular competence. Initial treatment is typically glucocorticoids at a dose of 1 mg/kg/day. Although the majority of patients will respond to this treatment, some cases are glucocorticoid resistant. In addition, relapse with tapering steroids is common. Most patients will require other immunosuppressive agents, which are often initiated at the time of diagnosis.8 An initial open-label study of methotrexate with glucocorticoids in 16 patients showed remission in 81% of patients and sustained remission in 50% of patients, suggesting its utility as a steroid-sparing agent.9 Limited data also exists to support the use of azathioprine, mycophenolate, cyclosporine, tacrolimus, leflunomide or cyclophosphamide.10 In all cases, a significant number of patients continue to have active disease or are unable to discontinue glucocorticoid treatment.