Innate Immunity Weighs In
For all the sophistication of adaptive immunity, with specificity and memory as its pinnacle achievements, it was recognized that the time required to expand the T-cell and B-cell responses in the setting of acute infection could place the host in great peril. The concept of innate immunity as the first line of host defense had been the province of leukocyte biology students, subsequently drosophila scholars. The discovery of toll-like receptors (TLRs) as the quintessential recognition elements of the innate immune system created an intense interest in defining the role of this family of receptors in infectious disease. However, the initial concepts proposed by the late Charles Janeway, MD, and colleagues seemed to have placed some conceptual hurdles in the path of linking TLR to rheumatic diseases.
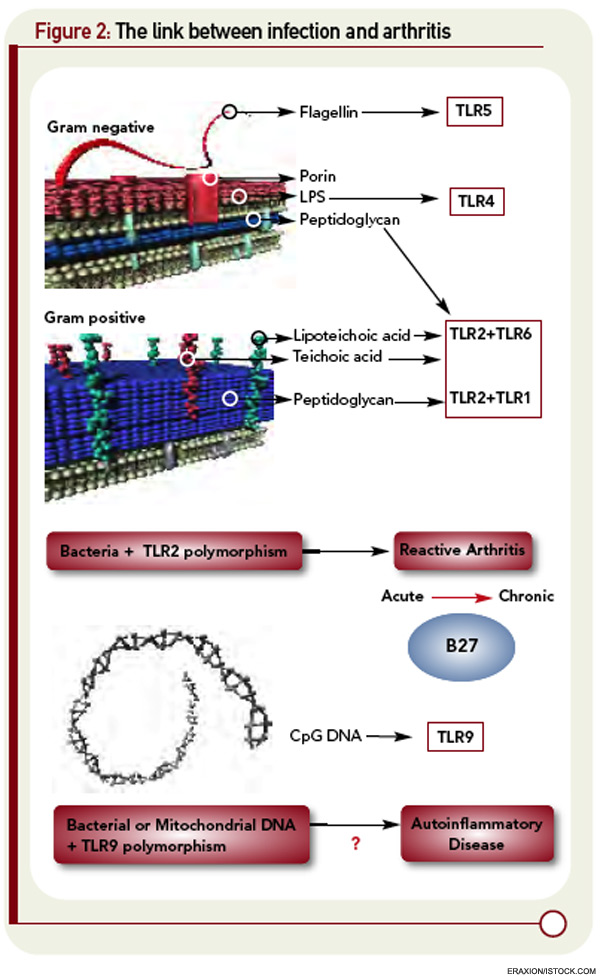
The first of these hurdles related to the apparent nonpolymorphic nature of the TLR, posing a significant challenge to addressing the heterogeneity of susceptibility to ReA in target populations. The second was that self/nonself discrimination by the innate immune system was proposed to be perfect, unlike its forever failing counterpart in adaptive immunity. Both of these concepts have needed significant revision. There are, in fact, significant polymorphisms in the TLR families, and recent studies in epidemic ReA have demonstrated that nonsynonymous single nucleotide polymorphisms in TLR2 confer susceptibility to post-Salmonella ReA (see Figure 2, p. 28).11 The notion that TLRs are specific only for foreign determinants has been replaced with the recognition that mammalian nucleic acids, among other molecules, are ligands for certain TLRs; indeed, this concept has proved to be pivotal in current concepts of lupus pathogenesis.