 ACR CONVERGENCE 2021—In a session on the diagnosis and treatment of interstitial lung disease (ILD) in patients with rheumatic diseases, Melissa Griffith, MD, assistant professor of medicine at the University of Colorado Anschutz Medical Campus, Aurora, Colo., began her presentation by reviewing common presentations of autoimmune lung disease.
ACR CONVERGENCE 2021—In a session on the diagnosis and treatment of interstitial lung disease (ILD) in patients with rheumatic diseases, Melissa Griffith, MD, assistant professor of medicine at the University of Colorado Anschutz Medical Campus, Aurora, Colo., began her presentation by reviewing common presentations of autoimmune lung disease.
“These are exceedingly rare diseases, with a paucity of trials and data,” said Dr. Griffith.
CTD-Associated ILD
“Patients with [ILD] may not yet have [an] identifiable rheumatic disease, but many eventually develop one,” said Dr. Griffith.
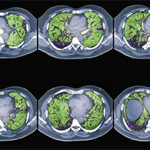
The most common patterns of ILD in connective tissue disease (CTD) are interstitial pneumonia, nonspecific interstitial pneumonia, organizing pneumonia and lymphocytic interstitial pneumonia.1 According to Dr. Griffith, these patterns are important because they guide prognosis. She emphasized that high-resolution computed tomography (CT) is crucial for diagnosing pulmonary fibrosis and should be used instead of traditional CT or chest radiographs.2
Diagnosis will likely require input from multiple healthcare providers. Dr. Griffith reinforced this point with data from a multidisciplinary ILD clinic at Massachusetts General Hospital, Boston. Researchers evaluated 50 patients referred to the tertiary care practice over a 12-month period. Patients were independently assessed by a rheumatologist and a pulmonologist. The investigators found the diagnosis was changed in 54% of patients who presented to the ILD clinic. Moreover, the evaluation resulted in changes in therapy for 80% of patients with CTD-associated ILD and 27% of patients with idiopathic pulmonary fibrosis. The authors concluded that rheumatologists should play a significant role in the clinical care of these patients and evaluate all patients with ILD.3
Rheumatologists should obtain autoimmune serologies on most patients with ILD, said Dr. Griffith, because this information can be useful in guiding therapy. Example: In the PANTHER-IPF trial, patients with idiopathic pulmonary fibrosis treated with a combination of prednisone, azathioprine and N-acetylcysteine were found to have higher risks of hospitalization and death than patients treated with placebo.4
“This [finding] led to a fundamental change in the way pulmonologists treat idiopathic pulmonary fibrosis,” explained Dr. Griffith.
Scleroderma-Associated ILD
In this section of her presentation, Dr. Griffith explained that ILD does not affect all patients with scleroderma equally. Although approximately half of patients with scleroderma will eventually be diagnosed with ILD, the risk for ILD varies by the type of scleroderma. For patients with scleroderma who are diagnosed with ILD, poor prognostic features include disease duration of less than four years, diffuse scleroderma, forced vital capacity (FVC) of <65%, diffusing capacity of carbon monoxide (DLCO) of <55%, a finding of more than 20% lung involvement as determined by high-resolution CT and SCL-70 antibody positivity.